–†–µ–Ј—Г–ї—М—В–∞—В—Л –Љ–∞—Б—Б–Њ–≤–Њ–≥–Њ —Б–Ї—А–Є–љ–Є–љ–≥–∞ –Є¬†–Ї–ї–Є–љ–Є–Ї–Њ-–њ–∞—А–∞–Ї–ї–Є–љ–Є—З–µ—Б–Ї–∞—П —Е–∞—А–∞–Ї—В–µ—А–Є—Б—В–Є–Ї–∞ –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†—В—А–∞–љ—Б—В–Є—А–µ—В–Є–љ–Њ–≤–Њ–є —Б–µ–Љ–µ–є–љ–Њ–є –∞–Љ–Є–ї–Њ–Є–і–љ–Њ–є –њ–Њ–ї–Є–љ–µ–є—А–Њ–њ–∞—В–Є–µ–є
- –Р–љ–љ–Њ—В–∞—Ж–Є—П
- –°—В–∞—В—М—П
- –°—Б—Л–ї–Ї–Є
- English
–¶–µ–ї—М –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П вАУ –њ—А–Њ–∞–љ–∞–ї–Є–Ј–Є—А–Њ–≤–∞—В—М —Б–ї—Г—З–∞–Є –Ґ–Ґ–†-–°–Р–Я, –≤—Л—П–≤–ї–µ–љ–љ—Л–µ –≤ –§–У–С–Э–£ ¬Ђ–Э–∞—Г—З–љ—Л–є —Ж–µ–љ—В—А –љ–µ–≤—А–Њ–ї–Њ–≥–Є–Є¬ї –≤ –њ–µ—А–Є–Њ–і 2018вАУ2025 –≥–≥.
–Ь–∞—В–µ—А–Є–∞–ї –Є –Љ–µ—В–Њ–і—Л. –° 2018 –њ–Њ 2025 –≥. –≤ –§–У–С–Э–£ ¬Ђ–Э–∞—Г—З–љ—Л–є —Ж–µ–љ—В—А –љ–µ–≤—А–Њ–ї–Њ–≥–Є–Є¬ї –њ—А–Њ–≤–µ–і–µ–љ —Б–Ї—А–Є–љ–Є–љ–≥ –љ–∞ –Ґ–Ґ–†-–°–Р–Я 750 –њ–∞—Ж–Є–µ–љ—В–∞–Љ —Б —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є –њ–Њ–ї–Є–љ–µ–є—А–Њ–њ–∞—В–Є–µ–є –љ–µ—Г—В–Њ—З–љ–µ–љ–љ–Њ–≥–Њ –≥–µ–љ–µ–Ј–∞ —Б –њ—А–Є–Љ–µ–љ–µ–љ–Є–µ–Љ –∞–ї–≥–Њ—А–Є—В–Љ–∞ –Њ—В–±–Њ—А–∞ –њ–Њ –Ї–ї—О—З–µ–≤—Л–Љ –і–Є–∞–≥–љ–Њ–Ј–∞–Љ –≤ –Ї–Њ–Љ–±–Є–љ–∞—Ж–Є–Є —Б ¬Ђ–Ї—А–∞—Б–љ—Л–Љ–Є —Д–ї–∞–ґ–Ї–∞–Љ–Є¬ї. –£ –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б –≤–µ—А–Є—Д–Є—Ж–Є—А–Њ–≤–∞–љ–љ—Л–Љ –і–Є–∞–≥–љ–Њ–Ј–Њ–Љ –Ґ–Ґ–†-–°–Р–Я –њ—А–Њ–∞–љ–∞–ї–Є–Ј–Є—А–Њ–≤–∞–љ—Л –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ, –Є–љ—Б—В—А—Г–Љ–µ–љ—В–∞–ї—М–љ—Л–µ –Є –ї–∞–±–Њ—А–∞—В–Њ—А–љ—Л–µ —Е–∞—А–∞–Ї—В–µ—А–Є—Б—В–Є–Ї–Є.
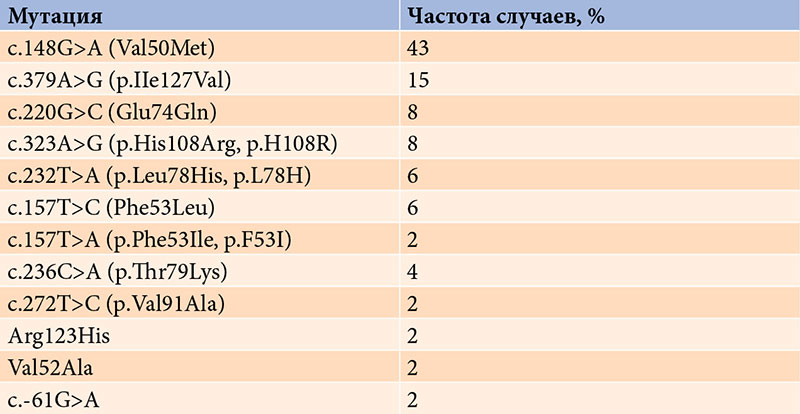
–†–µ–Ј—Г–ї—М—В–∞—В—Л. –Ь—Г—В–∞—Ж–Є—П –≤ –≥–µ–љ–µ TTR –±—Л–ї–∞ –≤—Л—П–≤–ї–µ–љ–∞ —Г 35 (5%) –±–Њ–ї—М–љ—Л—Е: —Г 21 (60%) –Љ—Г–ґ—З–Є–љ—Л –Є 14 (40%) –ґ–µ–љ—Й–Є–љ. –Ф–Њ–њ–Њ–ї–љ–Є—В–µ–ї—М–љ–Њ–µ –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ–Њ-–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Њ–µ –Њ–±—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ 24 —А–Њ–і—Б—В–≤–µ–љ–љ–Є–Ї–Њ–≤ –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –њ–Њ–Ј–≤–Њ–ї–Є–ї–Њ –≤—Л—П–≤–Є—В—М 13 –±–µ—Б—Б–Є–Љ–њ—В–Њ–Љ–љ—Л—Е –љ–Њ—Б–Є—В–µ–ї–µ–є –Љ—Г—В–∞–љ—В–љ–Њ–≥–Њ –≥–µ–љ–∞. –Э–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В–Њ–є –Њ–Ї–∞–Ј–∞–ї–∞—Б—М –Љ—Г—В–∞—Ж–Є—П c.148G>A (Val50Met) вАУ 20 (43%) —Б–ї—Г—З–∞–µ–≤.
–°—А–µ–і–љ–Є–є –≤–Њ–Ј—А–∞—Б—В –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б–Њ—Б—В–∞–≤–Є–ї 52 –≥–Њ–і–∞ (–Љ–Є–љ–Є–Љ—Г–Љ вАУ 23 –≥–Њ–і–∞, –Љ–∞–Ї—Б–Є–Љ—Г–Љ вАУ 81 –≥–Њ–і). –°—А–µ–і–љ–µ–µ –≤—А–µ–Љ—П –Њ—В –љ–∞—З–∞–ї–∞ –±–Њ–ї–µ–Ј–љ–Є –і–Њ —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–Є—П –і–Є–∞–≥–љ–Њ–Ј–∞ вАУ 2,8 –≥–Њ–і–∞. –£ –≤—Б–µ—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤ (35 (100%)) –Њ–±–љ–∞—А—Г–ґ–µ–љ—Л –њ–Њ–ї–Є–љ–µ–≤—А–Є—В–Є—З–µ—Б–Ї–Є–µ –љ–∞—А—Г—И–µ–љ–Є—П –≤ —Б–Њ—З–µ—В–∞–љ–Є–Є —Б —Б–Є–Љ–њ—В–Њ–Љ–∞–Љ–Є –њ–Њ—А–∞–ґ–µ–љ–Є—П –ґ–µ–ї—Г–і–Њ—З–љ–Њ-–Ї–Є—И–µ—З–љ–Њ–≥–Њ —В—А–∞–Ї—В–∞ (22 (63%)) –Є —Б–µ—А–і–µ—З–љ–Њ-—Б–Њ—Б—Г–і–Є—Б—В–Њ–є —Б–Є—Б—В–µ–Љ—Л (12 (34%)). –£ 11 (31%) –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –≤–Ј—П—В –∞–љ–∞–ї–Є–Ј –Ї—А–Њ–≤–Є –љ–∞ NT-proBNP. –Я–Њ–≤—Л—И–µ–љ–љ—Л–є —Г—А–Њ–≤–µ–љ—М (–±–Њ–ї–µ–µ 125 –њ–≥/–Љ–ї) –≤—Л—П–≤–ї–µ–љ —Г 6 (55%) –њ–∞—Ж–Є–µ–љ—В–Њ–≤. –Я—А–Є –њ—А–Њ–≤–µ–і–µ–љ–Є–Є —Н–ї–µ–Ї—В—А–Њ–љ–µ–є—А–Њ–Љ–Є–Њ–≥—А–∞—Д–Є–Є –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Є—Е –љ–µ—А–≤–Њ–≤ —Г 31 (89%) –њ–∞—Ж–Є–µ–љ—В–∞ —Б –Ґ–Ґ–†-–°–Р–Я –Њ–±–љ–∞—А—Г–ґ–µ–љ—Л –љ–µ–є—А–Њ—Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –њ—А–Є–Ј–љ–∞–Ї–Є –≥–µ–љ–µ—А–∞–ї–Є–Ј–Њ–≤–∞–љ–љ–Њ–≥–Њ —Б–Є–Љ–Љ–µ—В—А–Є—З–љ–Њ–≥–Њ —Б–µ–љ—Б–Њ–Љ–Њ—В–Њ—А–љ–Њ–≥–Њ –љ–µ–≤—А–∞–ї—М–љ–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П. –Т 27 (87%) —Б–ї—Г—З–∞—П—Е –Є–Ј 31 (89%) –≤—Л—П–≤–ї–µ–љ –∞–Ї—Б–Њ–љ–∞–ї—М–љ—Л–є —Е–∞—А–∞–Ї—В–µ—А –њ–Њ—А–∞–ґ–µ–љ–Є—П –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Є—Е –љ–µ—А–≤–Њ–≤, –≤ 4 (13%) —Б–ї—Г—З–∞—П—Е вАУ –і–µ–Љ–Є–µ–ї–Є–љ–Є–Ј–Є—А—Г—Й–Є–є. –Я—А–Є –њ—А–Њ–≤–µ–і–µ–љ–Є–Є —Г–ї—М—В—А–∞–Ј–≤—Г–Ї–Њ–≤–Њ–≥–Њ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Є—Е –љ–µ—А–≤–Њ–≤ –њ–Њ –њ—А–Њ—В–Њ–Ї–Њ–ї—Г Ultrasound Pattern Sum Score 9 (26%) –њ–∞—Ж–Є–µ–љ—В–∞–Љ —Г 3 (33%) –Є–Ј –љ–Є—Е —Б—Г–Љ–Љ–∞—А–љ—Л–є –±–∞–ї–ї —Б–Њ—Б—В–∞–≤–Є–ї –і–µ–≤—П—В—М –Є –±–Њ–ї–µ–µ, —З—В–Њ —Б–Њ–Њ—В–≤–µ—В—Б—В–≤—Г–µ—В –Є–Ј–Љ–µ–љ–µ–љ–Є—П–Љ, —Е–∞—А–∞–Ї—В–µ—А–љ—Л–Љ –і–ї—П —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є –≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ–Њ–є –і–µ–Љ–Є–µ–ї–Є–љ–Є–Ј–Є—А—Г—О—Й–µ–є –њ–Њ–ї–Є—А–∞–і–Є–Ї—Г–ї–Њ–љ–µ–є—А–Њ–њ–∞—В–Є–Є. –£ 6 (67%) –±–Њ–ї—М–љ—Л—Е –Є–Ј–Љ–µ–љ–µ–љ–Є—П –±—Л–ї–Є –Љ—П–≥–Ї–Є–Љ–Є –Є–ї–Є –≤–Њ–≤—Б–µ –Њ—В—Б—Г—В—Б—В–≤–Њ–≤–∞–ї–Є. –Ш–Ј 7 (20%) –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б –Ґ–Ґ–†-–°–Р–Я, –Ї–Њ—В–Њ—А—Л–Љ –±—Л–ї–Њ –њ—А–Њ–≤–µ–і–µ–љ–Њ –Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –Є–Ї—А–Њ–љ–Њ–ґ–љ–Њ–≥–Њ –љ–µ—А–≤–∞ –Є –њ–Њ–і–Ї–Њ–ґ–љ–Њ-–ґ–Є—А–Њ–≤–Њ–є –Ї–ї–µ—В—З–∞—В–Ї–Є, —В–Њ–ї—М–Ї–Њ —Г 4 (57%) –Њ—В–Љ–µ—З–µ–љ—Л –Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –њ—А–Є–Ј–љ–∞–Ї–Є –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞.
–Ч–∞–Ї–ї—О—З–µ–љ–Є–µ. –Т —Н–њ–Њ—Е—Г —Б–Њ–≤–µ—А—И–µ–љ—Б—В–≤–Њ–≤–∞–љ–Є—П —В–∞—А–≥–µ—В–љ–Њ–є –њ–∞—В–Њ–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Њ–є —В–µ—А–∞–њ–Є–Є –Ґ–Ґ–†-–°–Р–Я –∞–Ї—В—Г–∞–ї—М–љ–Њ—Б—В—М —Б–≤–Њ–µ–≤—А–µ–Љ–µ–љ–љ–Њ–≥–Њ –≤—Л—П–≤–ї–µ–љ–Є—П –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П —З—А–µ–Ј–≤—Л—З–∞–є–љ–Њ –≤—Л—Б–Њ–Ї–∞. –Я—А–Є –љ–∞–ї–Є—З–Є–Є ¬Ђ–Ї—А–∞—Б–љ—Л—Е —Д–ї–∞–ґ–Ї–Њ–≤¬ї —Г –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–µ–є —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є –њ–Њ–ї–Є–љ–µ–є—А–Њ–њ–∞—В–Є–µ–є –њ—А–Њ–≤–µ–і–µ–љ–Є–µ –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ–Њ-–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Њ–≥–Њ —Б–Ї—А–Є–љ–Є–љ–≥–∞ –љ–∞ –Ґ–Ґ–†-–°–Р–Я –њ–Њ–Ј–≤–Њ–ї—П–µ—В –њ–Њ–і—В–≤–µ—А–і–Є—В—М –і–Є–∞–≥–љ–Њ–Ј. –£ –±–Њ–ї—М—И–Є–љ—Б—В–≤–∞ —Б—В—А–∞–і–∞—О—Й–Є—Е –Ґ–Ґ–†-–°–Р–Я –Є–Љ–µ–µ—В –Љ–µ—Б—В–Њ –њ–Њ–Ј–і–љ–Є–є –і–µ–±—О—В. –Я—А–Є–Ј–љ–∞–Ї–Є –њ–Њ–ї–Є–љ–µ–є—А–Њ–њ–∞—В–Є–Є —Б–Њ—З–µ—В–∞—О—В—Б—П —Б –ґ–µ–ї—Г–і–Њ—З–љ–Њ-–Ї–Є—И–µ—З–љ—Л–Љ–Є, —Б–µ—А–і–µ—З–љ–Њ-—Б–Њ—Б—Г–і–Є—Б—В—Л–Љ–Є —Б–Є–Љ–њ—В–Њ–Љ–∞–Љ–Є –Є –љ–µ–њ—А–µ–і–љ–∞–Љ–µ—А–µ–љ–љ–Њ–є –њ–Њ—В–µ—А–µ–є –Љ–∞—Б—Б—Л —В–µ–ї–∞. –≠–ї–µ–Ї—В—А–Њ–љ–µ–є—А–Њ–Љ–Є–Њ–≥—А–∞—Д–Є—П –њ–Њ–і—В–≤–µ—А–ґ–і–∞–µ—В —Е–∞—А–∞–Ї—В–µ—А –њ–Њ—А–∞–ґ–µ–љ–Є—П –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Є—Е –љ–µ—А–≤–Њ–≤, —З–∞—Й–µ –≤—Б–µ–≥–Њ –њ–µ—А–≤–Є—З–љ–Њ –∞–Ї—Б–Њ–љ–∞–ї—М–љ—Л–є. –£–ї—М—В—А–∞–Ј–≤—Г–Ї–Њ–≤–Њ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Є—Е –љ–µ—А–≤–Њ–≤ –Є –±–Є–Њ–њ—Б–Є—П –Є–Ї—А–Њ–љ–Њ–ґ–љ–Њ–≥–Њ –љ–µ—А–≤–∞ –±–Њ–ї–µ–µ —З–µ–Љ –≤ —В—А–µ—В–Є —Б–ї—Г—З–∞–µ–≤ –љ–µ –і–∞—О—В –њ–Њ–ї–µ–Ј–љ–Њ–є –і–ї—П –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–Є –Ґ–Ґ–†-–°–Р–Я –Є–љ—Д–Њ—А–Љ–∞—Ж–Є–Є.
–¶–µ–ї—М –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П вАУ –њ—А–Њ–∞–љ–∞–ї–Є–Ј–Є—А–Њ–≤–∞—В—М —Б–ї—Г—З–∞–Є –Ґ–Ґ–†-–°–Р–Я, –≤—Л—П–≤–ї–µ–љ–љ—Л–µ –≤ –§–У–С–Э–£ ¬Ђ–Э–∞—Г—З–љ—Л–є —Ж–µ–љ—В—А –љ–µ–≤—А–Њ–ї–Њ–≥–Є–Є¬ї –≤ –њ–µ—А–Є–Њ–і 2018вАУ2025 –≥–≥.
–Ь–∞—В–µ—А–Є–∞–ї –Є –Љ–µ—В–Њ–і—Л. –° 2018 –њ–Њ 2025 –≥. –≤ –§–У–С–Э–£ ¬Ђ–Э–∞—Г—З–љ—Л–є —Ж–µ–љ—В—А –љ–µ–≤—А–Њ–ї–Њ–≥–Є–Є¬ї –њ—А–Њ–≤–µ–і–µ–љ —Б–Ї—А–Є–љ–Є–љ–≥ –љ–∞ –Ґ–Ґ–†-–°–Р–Я 750 –њ–∞—Ж–Є–µ–љ—В–∞–Љ —Б —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є –њ–Њ–ї–Є–љ–µ–є—А–Њ–њ–∞—В–Є–µ–є –љ–µ—Г—В–Њ—З–љ–µ–љ–љ–Њ–≥–Њ –≥–µ–љ–µ–Ј–∞ —Б –њ—А–Є–Љ–µ–љ–µ–љ–Є–µ–Љ –∞–ї–≥–Њ—А–Є—В–Љ–∞ –Њ—В–±–Њ—А–∞ –њ–Њ –Ї–ї—О—З–µ–≤—Л–Љ –і–Є–∞–≥–љ–Њ–Ј–∞–Љ –≤ –Ї–Њ–Љ–±–Є–љ–∞—Ж–Є–Є —Б ¬Ђ–Ї—А–∞—Б–љ—Л–Љ–Є —Д–ї–∞–ґ–Ї–∞–Љ–Є¬ї. –£ –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б –≤–µ—А–Є—Д–Є—Ж–Є—А–Њ–≤–∞–љ–љ—Л–Љ –і–Є–∞–≥–љ–Њ–Ј–Њ–Љ –Ґ–Ґ–†-–°–Р–Я –њ—А–Њ–∞–љ–∞–ї–Є–Ј–Є—А–Њ–≤–∞–љ—Л –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ, –Є–љ—Б—В—А—Г–Љ–µ–љ—В–∞–ї—М–љ—Л–µ –Є –ї–∞–±–Њ—А–∞—В–Њ—А–љ—Л–µ —Е–∞—А–∞–Ї—В–µ—А–Є—Б—В–Є–Ї–Є.
–†–µ–Ј—Г–ї—М—В–∞—В—Л. –Ь—Г—В–∞—Ж–Є—П –≤ –≥–µ–љ–µ TTR –±—Л–ї–∞ –≤—Л—П–≤–ї–µ–љ–∞ —Г 35 (5%) –±–Њ–ї—М–љ—Л—Е: —Г 21 (60%) –Љ—Г–ґ—З–Є–љ—Л –Є 14 (40%) –ґ–µ–љ—Й–Є–љ. –Ф–Њ–њ–Њ–ї–љ–Є—В–µ–ї—М–љ–Њ–µ –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ–Њ-–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Њ–µ –Њ–±—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ 24 —А–Њ–і—Б—В–≤–µ–љ–љ–Є–Ї–Њ–≤ –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –њ–Њ–Ј–≤–Њ–ї–Є–ї–Њ –≤—Л—П–≤–Є—В—М 13 –±–µ—Б—Б–Є–Љ–њ—В–Њ–Љ–љ—Л—Е –љ–Њ—Б–Є—В–µ–ї–µ–є –Љ—Г—В–∞–љ—В–љ–Њ–≥–Њ –≥–µ–љ–∞. –Э–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В–Њ–є –Њ–Ї–∞–Ј–∞–ї–∞—Б—М –Љ—Г—В–∞—Ж–Є—П c.148G>A (Val50Met) вАУ 20 (43%) —Б–ї—Г—З–∞–µ–≤.
–°—А–µ–і–љ–Є–є –≤–Њ–Ј—А–∞—Б—В –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б–Њ—Б—В–∞–≤–Є–ї 52 –≥–Њ–і–∞ (–Љ–Є–љ–Є–Љ—Г–Љ вАУ 23 –≥–Њ–і–∞, –Љ–∞–Ї—Б–Є–Љ—Г–Љ вАУ 81 –≥–Њ–і). –°—А–µ–і–љ–µ–µ –≤—А–µ–Љ—П –Њ—В –љ–∞—З–∞–ї–∞ –±–Њ–ї–µ–Ј–љ–Є –і–Њ —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–Є—П –і–Є–∞–≥–љ–Њ–Ј–∞ вАУ 2,8 –≥–Њ–і–∞. –£ –≤—Б–µ—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤ (35 (100%)) –Њ–±–љ–∞—А—Г–ґ–µ–љ—Л –њ–Њ–ї–Є–љ–µ–≤—А–Є—В–Є—З–µ—Б–Ї–Є–µ –љ–∞—А—Г—И–µ–љ–Є—П –≤ —Б–Њ—З–µ—В–∞–љ–Є–Є —Б —Б–Є–Љ–њ—В–Њ–Љ–∞–Љ–Є –њ–Њ—А–∞–ґ–µ–љ–Є—П –ґ–µ–ї—Г–і–Њ—З–љ–Њ-–Ї–Є—И–µ—З–љ–Њ–≥–Њ —В—А–∞–Ї—В–∞ (22 (63%)) –Є —Б–µ—А–і–µ—З–љ–Њ-—Б–Њ—Б—Г–і–Є—Б—В–Њ–є —Б–Є—Б—В–µ–Љ—Л (12 (34%)). –£ 11 (31%) –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –≤–Ј—П—В –∞–љ–∞–ї–Є–Ј –Ї—А–Њ–≤–Є –љ–∞ NT-proBNP. –Я–Њ–≤—Л—И–µ–љ–љ—Л–є —Г—А–Њ–≤–µ–љ—М (–±–Њ–ї–µ–µ 125 –њ–≥/–Љ–ї) –≤—Л—П–≤–ї–µ–љ —Г 6 (55%) –њ–∞—Ж–Є–µ–љ—В–Њ–≤. –Я—А–Є –њ—А–Њ–≤–µ–і–µ–љ–Є–Є —Н–ї–µ–Ї—В—А–Њ–љ–µ–є—А–Њ–Љ–Є–Њ–≥—А–∞—Д–Є–Є –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Є—Е –љ–µ—А–≤–Њ–≤ —Г 31 (89%) –њ–∞—Ж–Є–µ–љ—В–∞ —Б –Ґ–Ґ–†-–°–Р–Я –Њ–±–љ–∞—А—Г–ґ–µ–љ—Л –љ–µ–є—А–Њ—Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –њ—А–Є–Ј–љ–∞–Ї–Є –≥–µ–љ–µ—А–∞–ї–Є–Ј–Њ–≤–∞–љ–љ–Њ–≥–Њ —Б–Є–Љ–Љ–µ—В—А–Є—З–љ–Њ–≥–Њ —Б–µ–љ—Б–Њ–Љ–Њ—В–Њ—А–љ–Њ–≥–Њ –љ–µ–≤—А–∞–ї—М–љ–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П. –Т 27 (87%) —Б–ї—Г—З–∞—П—Е –Є–Ј 31 (89%) –≤—Л—П–≤–ї–µ–љ –∞–Ї—Б–Њ–љ–∞–ї—М–љ—Л–є —Е–∞—А–∞–Ї—В–µ—А –њ–Њ—А–∞–ґ–µ–љ–Є—П –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Є—Е –љ–µ—А–≤–Њ–≤, –≤ 4 (13%) —Б–ї—Г—З–∞—П—Е вАУ –і–µ–Љ–Є–µ–ї–Є–љ–Є–Ј–Є—А—Г—Й–Є–є. –Я—А–Є –њ—А–Њ–≤–µ–і–µ–љ–Є–Є —Г–ї—М—В—А–∞–Ј–≤—Г–Ї–Њ–≤–Њ–≥–Њ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Є—Е –љ–µ—А–≤–Њ–≤ –њ–Њ –њ—А–Њ—В–Њ–Ї–Њ–ї—Г Ultrasound Pattern Sum Score 9 (26%) –њ–∞—Ж–Є–µ–љ—В–∞–Љ —Г 3 (33%) –Є–Ј –љ–Є—Е —Б—Г–Љ–Љ–∞—А–љ—Л–є –±–∞–ї–ї —Б–Њ—Б—В–∞–≤–Є–ї –і–µ–≤—П—В—М –Є –±–Њ–ї–µ–µ, —З—В–Њ —Б–Њ–Њ—В–≤–µ—В—Б—В–≤—Г–µ—В –Є–Ј–Љ–µ–љ–µ–љ–Є—П–Љ, —Е–∞—А–∞–Ї—В–µ—А–љ—Л–Љ –і–ї—П —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є –≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ–Њ–є –і–µ–Љ–Є–µ–ї–Є–љ–Є–Ј–Є—А—Г—О—Й–µ–є –њ–Њ–ї–Є—А–∞–і–Є–Ї—Г–ї–Њ–љ–µ–є—А–Њ–њ–∞—В–Є–Є. –£ 6 (67%) –±–Њ–ї—М–љ—Л—Е –Є–Ј–Љ–µ–љ–µ–љ–Є—П –±—Л–ї–Є –Љ—П–≥–Ї–Є–Љ–Є –Є–ї–Є –≤–Њ–≤—Б–µ –Њ—В—Б—Г—В—Б—В–≤–Њ–≤–∞–ї–Є. –Ш–Ј 7 (20%) –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б –Ґ–Ґ–†-–°–Р–Я, –Ї–Њ—В–Њ—А—Л–Љ –±—Л–ї–Њ –њ—А–Њ–≤–µ–і–µ–љ–Њ –Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –Є–Ї—А–Њ–љ–Њ–ґ–љ–Њ–≥–Њ –љ–µ—А–≤–∞ –Є –њ–Њ–і–Ї–Њ–ґ–љ–Њ-–ґ–Є—А–Њ–≤–Њ–є –Ї–ї–µ—В—З–∞—В–Ї–Є, —В–Њ–ї—М–Ї–Њ —Г 4 (57%) –Њ—В–Љ–µ—З–µ–љ—Л –Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –њ—А–Є–Ј–љ–∞–Ї–Є –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞.
–Ч–∞–Ї–ї—О—З–µ–љ–Є–µ. –Т —Н–њ–Њ—Е—Г —Б–Њ–≤–µ—А—И–µ–љ—Б—В–≤–Њ–≤–∞–љ–Є—П —В–∞—А–≥–µ—В–љ–Њ–є –њ–∞—В–Њ–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Њ–є —В–µ—А–∞–њ–Є–Є –Ґ–Ґ–†-–°–Р–Я –∞–Ї—В—Г–∞–ї—М–љ–Њ—Б—В—М —Б–≤–Њ–µ–≤—А–µ–Љ–µ–љ–љ–Њ–≥–Њ –≤—Л—П–≤–ї–µ–љ–Є—П –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П —З—А–µ–Ј–≤—Л—З–∞–є–љ–Њ –≤—Л—Б–Њ–Ї–∞. –Я—А–Є –љ–∞–ї–Є—З–Є–Є ¬Ђ–Ї—А–∞—Б–љ—Л—Е —Д–ї–∞–ґ–Ї–Њ–≤¬ї —Г –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–µ–є —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є –њ–Њ–ї–Є–љ–µ–є—А–Њ–њ–∞—В–Є–µ–є –њ—А–Њ–≤–µ–і–µ–љ–Є–µ –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ–Њ-–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Њ–≥–Њ —Б–Ї—А–Є–љ–Є–љ–≥–∞ –љ–∞ –Ґ–Ґ–†-–°–Р–Я –њ–Њ–Ј–≤–Њ–ї—П–µ—В –њ–Њ–і—В–≤–µ—А–і–Є—В—М –і–Є–∞–≥–љ–Њ–Ј. –£ –±–Њ–ї—М—И–Є–љ—Б—В–≤–∞ —Б—В—А–∞–і–∞—О—Й–Є—Е –Ґ–Ґ–†-–°–Р–Я –Є–Љ–µ–µ—В –Љ–µ—Б—В–Њ –њ–Њ–Ј–і–љ–Є–є –і–µ–±—О—В. –Я—А–Є–Ј–љ–∞–Ї–Є –њ–Њ–ї–Є–љ–µ–є—А–Њ–њ–∞—В–Є–Є —Б–Њ—З–µ—В–∞—О—В—Б—П —Б –ґ–µ–ї—Г–і–Њ—З–љ–Њ-–Ї–Є—И–µ—З–љ—Л–Љ–Є, —Б–µ—А–і–µ—З–љ–Њ-—Б–Њ—Б—Г–і–Є—Б—В—Л–Љ–Є —Б–Є–Љ–њ—В–Њ–Љ–∞–Љ–Є –Є –љ–µ–њ—А–µ–і–љ–∞–Љ–µ—А–µ–љ–љ–Њ–є –њ–Њ—В–µ—А–µ–є –Љ–∞—Б—Б—Л —В–µ–ї–∞. –≠–ї–µ–Ї—В—А–Њ–љ–µ–є—А–Њ–Љ–Є–Њ–≥—А–∞—Д–Є—П –њ–Њ–і—В–≤–µ—А–ґ–і–∞–µ—В —Е–∞—А–∞–Ї—В–µ—А –њ–Њ—А–∞–ґ–µ–љ–Є—П –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Є—Е –љ–µ—А–≤–Њ–≤, —З–∞—Й–µ –≤—Б–µ–≥–Њ –њ–µ—А–≤–Є—З–љ–Њ –∞–Ї—Б–Њ–љ–∞–ї—М–љ—Л–є. –£–ї—М—В—А–∞–Ј–≤—Г–Ї–Њ–≤–Њ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Є—Е –љ–µ—А–≤–Њ–≤ –Є –±–Є–Њ–њ—Б–Є—П –Є–Ї—А–Њ–љ–Њ–ґ–љ–Њ–≥–Њ –љ–µ—А–≤–∞ –±–Њ–ї–µ–µ —З–µ–Љ –≤ —В—А–µ—В–Є —Б–ї—Г—З–∞–µ–≤ –љ–µ –і–∞—О—В –њ–Њ–ї–µ–Ј–љ–Њ–є –і–ї—П –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–Є –Ґ–Ґ–†-–°–Р–Я –Є–љ—Д–Њ—А–Љ–∞—Ж–Є–Є.
–Т–≤–µ–і–µ–љ–Є–µ
–Ґ—А–∞–љ—Б—В–Є—А–µ—В–Є–љ–Њ–≤–∞—П —Б–µ–Љ–µ–є–љ–∞—П –∞–Љ–Є–ї–Њ–Є–і–љ–∞—П –њ–Њ–ї–Є–љ–µ–є—А–Њ¬≠–њ–∞—В–Є—П (–Ґ–Ґ–†-–°–Р–Я)¬†вАУ —А–µ–і–Ї–Њ–µ, —В—П–ґ–µ–ї–Њ–µ, –љ–µ—Г–Ї–ї–Њ–љ–љ–Њ –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–µ–µ –Љ—Г–ї—М—В–Є—Б–Є—Б—В–µ–Љ–љ–Њ–µ –∞—Г—В–Њ—Б–Њ–Љ–љ–Њ-–і–Њ–Љ–Є–љ–∞–љ—В–љ–Њ–µ –љ–∞—Б–ї–µ–і—Б—В–≤–µ–љ–љ–Њ–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ, –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–љ–Њ–µ –Љ—Г—В–∞—Ж–Є–µ–є –≤ –≥–µ–љ–µ —В—А–∞–љ—Б—В–Є—А–µ—В–Є–љ–∞ (TTR), –њ—А–Є–≤–Њ–і—П—Й–µ–є –Ї¬†–і–µ—Б—В–∞–±–Є–ї–Є–Ј–∞—Ж–Є–Є —В–µ—В—А–∞–Љ–µ—А–љ–Њ–є —Б—В—А—Г–Ї—В—Г—А—Л –±–µ–ї–Ї–∞ TTR¬†[1]. –Ъ–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ –њ—А–Њ—П–≤–ї–µ–љ–Є—П –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П —Б–≤—П–Ј–∞–љ—Л —Б¬†–≤–љ–µ–Ї–ї–µ—В–Њ—З–љ—Л–Љ –Њ—В–ї–Њ–ґ–µ–љ–Є–µ–Љ –њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е –∞–Љ–Є–ї–Њ–Є–і–љ—Л—Е —Д–Є–±—А–Є–ї–ї –≤¬†—А–∞–Ј–ї–Є—З–љ—Л—Е –Њ—А–≥–∞–љ–∞—Е –Є¬†—В–Ї–∞–љ—П—Е, –њ—А–Є —Н—В–Њ–Љ —З–∞—Й–µ –≤¬†–њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Є—Е –љ–µ—А–≤–∞—Е (–Ґ–Ґ–†-–°–Р–Я), —Б–µ—А–і—Ж–µ (—В—А–∞–љ—Б—В–Є—А–µ—В–Є–љ–Њ–≤–∞—П –Ї–∞—А–і–Є–Њ–Љ–Є–Њ–њ–∞—В–Є—П), –∞¬†—В–∞–Ї–ґ–µ –≤¬†–ґ–µ–ї—Г–і–Њ—З–љ–Њ-–Ї–Є—И–µ—З–љ–Њ–Љ —В—А–∞–Ї—В–µ, –њ–Њ—З–Ї–∞—Е, –≥–ї–∞–Ј–∞—Е, —А–µ–ґ–µ¬†вАУ –≤¬†–Њ–±–Њ–ї–Њ—З–Ї–∞—Е –≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ (–ї–µ–њ—В–Њ–Љ–µ–љ–Є–љ–≥–µ–∞–ї—М–љ—Л–є –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј) [2, 3]. –≠–љ–і–µ–Љ–Є—З–љ—Л–Љ–Є —Б—В—А–∞–љ–∞–Љ–Є –і–ї—П –і–∞–љ–љ–Њ–≥–Њ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П —Б—З–Є—В–∞—О—В—Б—П –Я–Њ—А—В—Г–≥–∞–ї–Є—П, –®–≤–µ—Ж–Є—П –Є¬†–ѓ–њ–Њ–љ–Є—П [4, 5]. –Ю–і–љ–∞–Ї–Њ –≤–Њ –≤—Б–µ–Љ –Љ–Є—А–µ –љ–∞–±–ї—О–і–∞–µ—В—Б—П —А–Њ—Б—В —З–Є—Б–ї–∞ –≤—Л—П–≤–ї–µ–љ–љ—Л—Е —Б–ї—Г—З–∞–µ–≤, –љ–µ—А–µ–і–Ї–Њ –≤–Њ–Ј–љ–Є–Ї–∞—О—Й–Є—Е —Г¬†–≤–Ј—А–Њ—Б–ї—Л—Е –Є¬†–љ–Њ—Б—П—Й–Є—Е —Б–њ–Њ—А–∞–і–Є—З–µ—Б–Ї–Є–є —Е–∞—А–∞–Ї—В–µ—А, —З—В–Њ, –≤–µ—А–Њ—П—В–љ–Њ, —Б–≤—П–Ј–∞–љ–Њ —Б¬†–ї—Г—З—И–µ–є –Њ—Б–≤–µ–і–Њ–Љ–ї–µ–љ–љ–Њ—Б—В—М—О –Њ¬†–Ґ–Ґ–†-–°–Р–Я –Є¬†–±–Њ–ї–µ–µ —И–Є—А–Њ–Ї–Є–Љ –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є–µ–Љ –≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Њ–≥–Њ —В–µ—Б—В–Є—А–Њ–≤–∞–љ–Є—П [6].
–Э–µ—Б–Ї–Њ–ї—М–Ї–Њ –і–µ—Б—П—В–Є–ї–µ—В–Є–є –љ–∞–Ј–∞–і –Ґ–Ґ–†-–°–Р–Я —Б—З–Є—В–∞–ї–∞—Б—М –љ–µ–Є–Ј–ї–µ—З–Є–Љ—Л–Љ —Д–∞—В–∞–ї—М–љ—Л–Љ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ–Љ. –Ю–і–љ–∞–Ї–Њ –≤¬†–љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П —А–∞–Ј—А–∞–±–Њ—В–∞–љ–∞ —Н—Д—Д–µ–Ї—В–Є–≤–љ–∞—П –њ–∞—В–Њ–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–∞—П —В–µ—А–∞–њ–Є—П –Ґ–Ґ–†-–°–Р–Я, –≤¬†—Б–≤—П–Ј–Є —Б¬†—З–µ–Љ –∞–Ї—В—Г–∞–ї—М–љ—Л–Љ —Б—В–∞–љ–Њ–≤–Є—В—Б—П –µ–µ –≤—Л—П–≤–ї–µ–љ–Є–µ –љ–∞¬†—А–∞–љ–љ–µ–є —Б—В–∞–і–Є–Є [7, 8].
–¶–µ–ї—М –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П
–¶–µ–ї—М—О –љ–∞—Б—В–Њ—П—Й–µ–≥–Њ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П —Б—В–∞–ї –∞–љ–∞–ї–Є–Ј —Б–ї—Г—З–∞–µ–≤ –Ґ–Ґ–†-–°–Р–Я, –≤—Л—П–≤–ї–µ–љ–љ—Л—Е –≤¬†–§–У–С–Э–£ ¬Ђ–Э–∞—Г—З–љ—Л–є —Ж–µ–љ—В—А –љ–µ–≤—А–Њ–ї–Њ–≥–Є–Є¬ї –≤¬†–њ–µ—А–Є–Њ–і —Б 2018 –њ–Њ 2025 –≥.
–Ь–∞—В–µ—А–Є–∞–ї –Є¬†–Љ–µ—В–Њ–і—Л
–°¬†2018 –њ–Њ¬†2025 –≥.¬†–≤ –§–У–С–Э–£ ¬Ђ–Э–∞—Г—З–љ—Л–є —Ж–µ–љ—В—А –љ–µ–≤—А–Њ–ї–Њ–≥–Є–Є¬ї –њ—А–Њ–≤–Њ–і–Є–ї—Б—П –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ–Њ-–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є–є —Б–Ї—А–Є–љ–Є–љ–≥ –љ–∞¬†–Љ—Г—В–∞—Ж–Є–Є –≥–µ–љ–∞ TTR 750 –њ–∞—Ж–Є–µ–љ—В–∞–Љ —Б¬†—Е—А–Њ–љ–Є—З–µ—Б¬≠–Ї–Њ–є –њ–Њ–ї–Є–љ–µ–є—А–Њ–њ–∞—В–Є–µ–є (–Я–Э–Я) –љ–µ—Г—В–Њ—З–љ–µ–љ–љ–Њ–≥–Њ –≥–µ–љ–µ–Ј–∞.
–Ъ—А–Є—В–µ—А–Є–Є –Њ—В–±–Њ—А–∞ –љ–∞¬†—Б–Ї—А–Є–љ–Є–љ–≥ (–Ї—А–Є—В–µ—А–Є–Є –≤–Ї–ї—О—З–µ–љ–Є—П):
- —Е—А–Њ–љ–Є—З–µ—Б–Ї–∞—П –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–∞—П —Б–µ–љ—Б–Њ—А–љ–∞—П, —Б–µ–љ—Б–Њ–Љ–Њ—В–Њ—А–љ–∞—П –Є–ї–Є —Б–µ–љ—Б–Њ–≤–µ–≥–µ—В–∞—В–Є–≤–љ–∞—П –∞–Ї—Б–Њ–љ–∞–ї—М–љ–∞—П –Я–Э–Я –љ–µ—Г—В–Њ—З–љ–µ–љ–љ–Њ–≥–Њ –≥–µ–љ–µ–Ј–∞;
- —В–Њ–љ–Ї–Њ–≤–Њ–ї–Њ–Ї–Њ–љ–љ–∞—П –±–Њ–ї–µ–≤–∞—П –Я–Э–Я –љ–µ—Г—В–Њ—З–љ–µ–љ–љ–Њ–≥–Њ –≥–µ–љ–µ–Ј–∞;
- —А–µ—Д—А–∞–Ї—В–µ—А–љ–∞—П –Ї¬†–њ–∞—В–Њ–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Њ–є —В–µ—А–∞–њ–Є–Є –њ–µ—А–≤–Њ–є –ї–Є–љ–Є–Є —Е—А–Њ–љ–Є—З–µ—Б–Ї–∞—П –≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ–∞—П –і–µ–Љ–Є–µ–ї–Є–љ–Є–Ј–Є—А—Г—О—Й–∞—П –њ–Њ–ї–Є—А–∞–і–Є–Ї—Г–ї–Њ–љ–µ–є—А–Њ–њ–∞—В–Є—П (–•–Т–Ф–Я);
- —Е—А–Њ–љ–Є—З–µ—Б–Ї–∞—П –Я–Э–Я –љ–µ—Г—В–Њ—З–љ–µ–љ–љ–Њ–≥–Њ –≥–µ–љ–µ–Ј–∞ —Б¬†—Б–Њ–њ—Г—В—Б—В–≤—Г—О—Й–Є–Љ –Ї–∞—А–њ–∞–ї—М–љ—Л–Љ —В—Г–љ–љ–µ–ї—М–љ—Л–Љ —Б–Є–љ–і—А–Њ–Љ–Њ–Љ.
–Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –њ—А–Є–Љ–µ–љ—П–ї–∞—Б—М —Б–Є—Б—В–µ–Љ–∞ ¬Ђ–Ї—А–∞—Б–љ—Л—Е —Д–ї–∞–ґ–Ї–Њ–≤¬ї, —А–µ–Ї–Њ–Љ–µ–љ–і–Њ–≤–∞–љ–љ–∞—П –Љ–µ–ґ–і—Г–љ–∞—А–Њ–і–љ—Л–Љ —Б–Њ–Њ–±—Й–µ—Б—В–≤–Њ–Љ –і–ї—П –Њ—В–±–Њ—А–∞ –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –љ–∞ –≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є–є —Б–Ї—А–Є–љ–Є–љ–≥ –љ–∞¬†–Ґ–Ґ–†-–°–Р–Я. –Т¬†–і–Њ–њ–Њ–ї–љ–µ–љ–Є–µ –Ї¬†–≤—Л—И–µ–Њ–њ–Є—Б–∞–љ¬≠–љ—Л–Љ –і–Є–∞–≥–љ–Њ–Ј–∞–Љ —Г—З–Є—В—Л–≤–∞–ї–Њ—Б—М –љ–∞–ї–Є—З–Є–µ –ґ–µ–ї—Г–і–Њ—З–љ–Њ-–Ї–Є—И–µ—З–љ—Л—Е —А–∞—Б—Б—В—А–Њ–є—Б—В–≤ (–Ј–∞–њ–Њ—А—Л, –і–Є–∞—А–µ—П –Є–ї–Є –Є—Е —Б–Њ—З–µ—В–∞–љ–Є–µ), —Б–µ—А–і–µ—З–љ–Њ-—Б–Њ—Б—Г–і–Є—Б—В—Л—Е –љ–∞—А—Г—И–µ–љ–Є–є (–±–ї–Њ–Ї–∞–і–∞ –≤–љ—Г—В—А–Є—Б–µ—А–і–µ—З–љ–Њ–є –њ—А–Њ–≤–Њ–і–Є–Љ–Њ—Б—В–Є, –Љ–µ—А—Ж–∞—В–µ–ї—М–љ–∞—П –∞—А–Є—В–Љ–Є—П –љ–Њ—А–Љ–Њ—Б–Є—Б—В–Њ–ї–Є—З–µ—Б–Ї–Њ–є —Д–Њ—А–Љ—Л, —Е—А–Њ–љ–Є—З–µ—Б–Ї–∞—П —Б–µ—А–і–µ—З–љ–∞—П –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М —Б¬†—Б–Њ—Е—А–∞–љ–µ–љ–љ–Њ–є —Д—А–∞–Ї—Ж–Є–µ–є –≤—Л–±—А–Њ—Б–∞, –Ї–∞—А–і–Є–Њ–Љ–Є–Њ–њ–∞—В–Є—П, –Њ—А—В–Њ—Б—В–∞—В–Є—З–µ—Б–Ї–∞—П –≥–Є–њ–Њ—В–µ–љ–Ј–Є—П, –ї–Є–њ–Њ—В–Є–Љ–Є—П), –Ї–∞—А–њ–∞–ї—М–љ–Њ–≥–Њ —В—Г–љ–љ–µ–ї—М–љ–Њ–≥–Њ —Б–Є–љ–і—А–Њ–Љ–∞, –љ–∞—Б–ї–µ–і—Б—В–≤–µ–љ–љ–Њ–≥–Њ –∞–љ–∞–Љ–љ–µ–Ј–∞ –≤¬†–Њ—В–љ–Њ—И–µ–љ–Є–Є –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞, —Б–µ—А–і–µ—З–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є, –њ–Њ–ї–Є–љ–µ–є—А–Њ¬≠–њ–∞—В–Є–Є¬†[2].
–Ь–Њ–ї–µ–Ї—Г–ї—П—А–љ–Њ-–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є–є —Б–Ї—А–Є–љ–Є–љ–≥ –њ—А–Њ–≤–Њ–і–Є–ї—Б—П —Б¬†–Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є–µ–Љ –Љ–µ—В–Њ–і–∞ ¬Ђ—Б—Г—Е–Њ–≥–Њ –њ—П—В–љ–∞¬ї (—Б–µ–Ї–≤–µ–љ–Є—А–Њ–≤–∞–љ–Є–µ –њ–Њ¬†–Љ–µ—В–Њ–і—Г F. Sanger). –Р–љ–∞–ї–Є–Ј –Њ—Б—Г—Й–µ—Б—В–≤–ї—П–ї–Є —Б–Њ—В—А—Г–і–љ–Є–Ї–Є –§–У–С–Э–£ ¬Ђ–Ь–µ–і–Є–Ї–Њ-–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є–є –љ–∞—Г—З–љ—Л–є —Ж–µ–љ—В—А –Є–Љ. –∞–Ї–∞–і–µ–Љ–Є–Ї–∞ –Э.–Я. –С–Њ—З–Ї–Њ–≤–∞¬ї.
–Ь–Њ–ї–µ–Ї—Г–ї—П—А–љ–Њ-–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є–є —Б–Ї—А–Є–љ–Є–љ–≥ —В–∞–Ї–ґ–µ –њ—А–Њ–≤–Њ–і–Є–ї—Б—П —А–Њ–і—Б—В–≤–µ–љ–љ–Є–Ї–∞–Љ –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–њ–Њ–і—В–≤–µ—А–ґ–і–µ–љ–љ–Њ–є –Ґ–Ґ–†-–°–Р–Я.
–Т¬†–Њ—В–љ–Њ—И–µ–љ–Є–Є –≤—Б–µ—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–Ґ–Ґ–†-–°–Р–Я –≤—Л–њ–Њ–ї–љ–µ–љ—Л —Б–±–Њ—А –Є¬†–∞–љ–∞–ї–Є–Ј –ґ–∞–ї–Њ–±, –∞¬†—В–∞–Ї–ґ–µ –і–∞–љ–љ—Л—Е –∞–љ–∞–Љ–љ–µ–Ј–∞ –ґ–Є–Ј–љ–Є –Є¬†–Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П, –Њ–±—Й–Є–є –Є¬†–љ–µ–≤—А–Њ–ї–Њ–≥–Є—З–µ—Б¬≠–Ї–Є–є –Њ—Б–Љ–Њ—В—А —Б¬†–Њ—Ж–µ–љ–Ї–Њ–є —Б–Є–ї—Л –Љ—Л—И—Ж –Ї–Њ–љ–µ—З–љ–Њ—Б—В–µ–є –њ–Њ¬†Medical Research Council sum score¬† (MRCss), —Н–ї–µ–Ї—В—А–Њ–љ–µ–є—А–Њ¬≠–Љ–Є–Њ–≥—А–∞—Д–Є—П (–≠–Э–Ь–У) —Б¬†–Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ–Љ –њ—А–Њ–≤–Њ–і—П—Й–µ–є —Д—Г–љ–Ї—Ж–Є–Є –і–ї–Є–љ–љ—Л—Е –љ–µ—А–≤–Њ–≤ –Ї–Њ–љ–µ—З–љ–Њ—Б—В–µ–є –Є¬†–Њ–±—Й–µ–њ—А–Є–љ—П—В—Л—Е –њ–∞—А–∞–Љ–µ—В—А–Њ–≤. –Э–µ–Ї–Њ—В–Њ—А—Л–Љ¬† –њ–∞—Ж–Є–µ–љ—В–∞–Љ (8 (23%)) –њ—А–Њ–≤–Њ–і–Є–ї–Њ—Б—М —Г–ї—М—В—А–∞–Ј–≤—Г–Ї–Њ–≤–Њ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Є—Е –љ–µ—А–≤–Њ–≤ –њ–Њ¬†–њ—А–Њ—В–Њ–Ї–Њ–ї—Г Ultrasound Pattern Sum Score (UPSS), —Б–Њ–≥–ї–∞—Б–љ–Њ –Ї–Њ—В–Њ—А–Њ–Љ—Г –Њ—Ж–µ–љ–Є–≤–∞–ї–∞—Б—М –њ–ї–Њ—Й–∞–і—М –њ–Њ–њ–µ—А–µ—З–љ–Њ–≥–Њ —Б–µ—З–µ–љ–Є—П –љ–µ—А–≤–Њ–≤ –Є¬†–Є–љ—В—А–∞–љ–µ–≤—А–∞–ї—М–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –њ–Њ¬†–Ї–ї–∞—Б—Б–Є—Д–Є–Ї–∞—Ж–Є–Є L.¬†Pauda –≤¬†12¬†—В–Њ—З–Ї–∞—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П —Б¬†–Њ–і–љ–Њ–є —Б—В–Њ—А–Њ–љ—Л [9]. –°—В–∞–і–Є—П –Ґ–Ґ–†-–°–Р–Я –Њ–њ—А–µ–і–µ–ї—П–ї–∞—Б—М –њ–Њ¬†—И–Ї–∞–ї–µ —В—А–∞–љ—Б—В–Є—А–µ—В–Є–љ–Њ–≤–Њ–є —Б–µ–Љ–µ–є–љ–Њ–є –∞–Љ–Є–ї–Њ–Є–і–љ–Њ–є –њ–Њ–ї–Є–љ–µ–є—А–Њ–њ–∞—В–Є–Є (Stages¬†of¬†Familial Amyloid Polyneuropathy for Locomotion) [10].
–Ю—В–і–µ–ї—М–љ—Л–Љ –њ–∞—Ж–Є–µ–љ—В–∞–Љ (7 (20%)) –±—Л–ї–Њ –њ—А–Њ–≤–µ–і–µ–љ–Њ –Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –Є–Ї—А–Њ–љ–Њ–ґ–љ–Њ–≥–Њ –љ–µ—А–≤–∞, 3 (43%) –Є–Ј¬†–љ–Є—Е —В–∞–Ї–ґ–µ –±—Л–ї–Њ –њ—А–Њ–≤–µ–і–µ–љ–Њ –Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –њ–Њ–і–Ї–Њ–ґ–љ–Њ-–ґ–Є—А–Њ–≤–Њ–є –Ї–ї–µ—В—З–∞—В–Ї–Є –Њ–Ї–Њ–ї–Њ–њ—Г–њ–Њ—З–љ–Њ–є –Њ–±–ї–∞—Б—В–Є.
–Т¬†—Е–Њ–і–µ –Њ–±—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –±—Л–ї–Є –Є—Б–Ї–ї—О—З–µ–љ—Л –Є–љ—Л–µ –њ—А–Є—З–Є–љ—Л –Я–Э–Я (–Ї—А–Є—В–µ—А–Є–Є –љ–µ–≤–Ї–ї—О—З–µ–љ–Є—П):
- —Е—А–Њ–љ–Є—З–µ—Б–Ї–Є–µ –і–Є–Ј–Є–Љ–Љ—Г–љ–љ—Л–µ –љ–µ–є—А–Њ–њ–∞—В–Є–Є;
- —Е—А–Њ–љ–Є—З–µ—Б–Ї–Є–µ —Б–Њ–Љ–∞—В–Є—З–µ—Б–Ї–Є–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П;
- –∞–ї–Ї–Њ–≥–Њ–ї–Є–Ј–Љ;
- –і–µ—Д–Є—Ж–Є—В –≤–Є—В–∞–Љ–Є–љ–Њ–≤ –≥—А—Г–њ–њ—Л –Т;
- –њ—А–Є–µ–Љ —В–Њ–Ї—Б–Є—З–љ—Л—Е –ї–µ–Ї–∞—А—Б—В–≤–µ–љ–љ—Л—Е –њ—А–µ–њ–∞—А–∞—В–Њ–≤.
–†–µ–Ј—Г–ї—М—В–∞—В—Л
–Ш–Ј¬†750 –њ–∞—Ж–Є–µ–љ—В–Њ–≤, –Ї–Њ—В–Њ—А—Л–µ —Б–Њ–Њ—В–≤–µ—В—Б—В–≤–Њ–≤–∞–ї–Є –Ї—А–Є—В–µ—А–Є—П–Љ –≤–Ї–ї—О—З–µ–љ–Є—П –Є¬†–Ї–Њ—В–Њ—А—Л–Љ –±—Л–ї –њ—А–Њ–≤–µ–і–µ–љ –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ–Њ-–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є–є —Б–Ї—А–Є–љ–Є–љ–≥, –Љ—Г—В–∞—Ж–Є—П –≤ –≥–µ–љ–µ TTR –±—Л–ї–∞ –≤—Л—П–≤–ї–µ–љ–∞ —Г¬†35 (5%): —Г¬†21 (60%) –Љ—Г–ґ—З–Є–љ—Л –Є¬†14¬†(40%) –ґ–µ–љ—Й–Є–љ. –°—А–µ–і–љ–Є–є –≤–Њ–Ј—А–∞—Б—В –±–Њ–ї—М–љ—Л—Е —Б–Њ—Б—В–∞–≤–Є–ї 52 –≥–Њ–і–∞ (–Љ–Є–љ–Є–Љ—Г–Љ¬†вАУ 23 –≥–Њ–і–∞, –Љ–∞–Ї—Б–Є–Љ—Г–Љ¬†вАУ 81 –≥–Њ–і).
–Ф–Њ–њ–Њ–ї–љ–Є—В–µ–ї—М–љ–Њ–µ —Ж–µ–ї–µ–љ–∞–њ—А–∞–≤–ї–µ–љ–љ–Њ–µ –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ–Њ-–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Њ–µ –Њ–±—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ 24 –±–ї–Є–ґ–∞–є—И–Є—Е –њ—А—П–Љ—Л—Е —А–Њ–і—Б—В–≤–µ–љ–љ–Є–Ї–Њ–≤ –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –њ–Њ–Ј–≤–Њ–ї–Є–ї–Њ –≤—Л—П–≤–Є—В—М 13¬†–љ–Њ—Б–Є—В–µ–ї–µ–є –Љ—Г—В–∞—Ж–Є–є –≤ –≥–µ–љ–µ TTR, —Г¬†–Ї–Њ—В–Њ—А—Л—Е –Њ—В—Б—Г—В—Б—В–≤–Њ–≤–∞–ї–Є –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ –њ—А–Є–Ј–љ–∞–Ї–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П (–±–µ—Б—Б–Є–Љ–њ—В–Њ–Љ–љ—Л–µ –љ–Њ—Б–Є—В–µ–ї–Є –Љ—Г—В–∞–љ—В–љ–Њ–≥–Њ –≥–µ–љ–∞). –Э–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В–Њ–є –Љ—Г—В–∞—Ж–Є–µ–є –≥–µ–љ–∞ TTR, –≤—Л—П–≤–ї–µ–љ–љ–Њ–є —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ –Є¬†–Є—Е —А–Њ–і—Б—В–≤–µ–љ–љ–Є–Ї–Њ–≤, –Њ–Ї–∞–Ј–∞–ї–∞—Б—М –Љ—Г—В–∞—Ж–Є—П c.148G>A (Val50Met)¬†вАУ 20 (43%) —Б–ї—Г—З–∞–µ–≤ (—В–∞–±–ї–Є—Ж–∞).
–Э–∞¬†–Њ—Б–љ–Њ–≤–∞–љ–Є–Є —А–µ–Ј—Г–ї—М—В–∞—В–Њ–≤ –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ–Њ-–≥–µ–љ–µ—В–Є—З–µ—Б¬≠–Ї–Њ–≥–Њ –Њ–±—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –љ–∞¬†–±–∞–Ј–µ –§–У–С–Э–£ ¬Ђ–Э–∞—Г—З–љ—Л–є —Ж–µ–љ—В—А –љ–µ–≤—А–Њ–ї–Њ–≥–Є–Є¬ї –±—Л–ї–∞ —Б—Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–∞ –±–∞–Ј–∞ –і–∞–љ–љ—Л—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–Ґ–Ґ–†-–°–Р–Я, –≤¬†–Ї–Њ—В–Њ—А—Г—О –≤–Њ—И–ї–Є –љ–Њ—Б–Є—В–µ–ї–Є –Љ—Г—В–∞—Ж–Є–є –≥–µ–љ–∞ TTR (n = 48), –Ї–∞–Ї —Б–Є–Љ–њ—В–Њ–Љ–љ—Л–µ (n = 35), —В–∞–Ї –Є¬†–±–µ—Б—Б–Є–Љ–њ—В–Њ–Љ–љ—Л–µ (n = 13). –£¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–Ї–ї–Є–љ–Є—З–µ—Б¬≠–Ї–Є–Љ–Є –њ—А–Њ—П–≤–ї–µ–љ–Є—П–Љ–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П (n = 35) –њ—А–Њ–∞–љ–∞–ї–Є–Ј–Є—А–Њ–≤–∞–љ—Л –Ї–ї–Є–љ–Є–Ї–Њ-–њ–∞—А–∞–Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ –і–∞–љ–љ—Л–µ.
–•–∞—А–∞–Ї—В–µ—А–Є—Б—В–Є–Ї–∞ –∞–љ–∞–Љ–љ–µ—Б—В–Є—З–µ—Б–Ї–Є—Е –і–∞–љ–љ—Л—Е
–Т¬†–∞–љ–∞–ї–Є–Ј–Є—А—Г–µ–Љ–Њ–є –≤—Л–±–Њ—А–Ї–µ (n = 35) —Б—А–µ–і–љ–µ–µ –≤—А–µ–Љ—П –Њ—В¬†–љ–∞—З–∞–ї–∞ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –і–Њ —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–Є—П –і–Є–∞–≥–љ–Њ–Ј–∞ —Б–Њ—Б—В–∞–≤–Є–ї–Њ 2,8 –≥–Њ–і–∞, –≤¬†—В–Њ –≤—А–µ–Љ—П –Ї–∞–Ї —Б—А–µ–і–љ–µ–µ –≤—А–µ–Љ—П –Њ—В¬†–Њ–±—А–∞—Й–µ–љ–Є—П –≤¬†–§–У–С–Э–£ ¬Ђ–Э–∞—Г—З–љ—Л–є —Ж–µ–љ—В—А –љ–µ–≤—А–Њ–ї–Њ–≥–Є–Є¬ї –і–Њ —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–Є—П –Њ–Ї–Њ–љ—З–∞—В–µ–ї—М–љ–Њ–≥–Њ –і–Є–∞–≥–љ–Њ–Ј–∞ –њ—А–Є –≤—Л—Б–Њ–Ї–Њ–є –љ–∞—Б—В–Њ—А–Њ–ґ–µ–љ–љ–Њ—Б—В–Є –≤¬†–Њ—В–љ–Њ—И–µ–љ–Є–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –≤¬†—Б—А–µ–і–љ–µ–Љ —Б–Њ—Б—В–∞–≤–Є–ї–Њ —В—А–Є –Љ–µ—Б—П—Ж–∞. –†–µ—В—А–Њ—Б–њ–µ–Ї—В–Є–≤–љ—Л–є –∞–љ–∞–ї–Є–Ј –∞–љ–∞–Љ–љ–µ—Б—В–Є—З–µ—Б–Ї–Є—Е –і–∞–љ–љ—Л—Е –њ–Њ–Ї–∞–Ј–∞–ї, —З—В–Њ –≤¬†–і–µ–±—О—В–µ –±–Њ–ї–µ–Ј–љ–Є —Г¬†–≤—Б–µ—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –њ–µ—А–≤—Л–Љ–Є —Б–Є–Љ–њ—В–Њ–Љ–∞–Љ–Є –±—Л–ї–Є —Б–µ–љ—Б–Њ—А–љ—Л–µ –њ–Њ–ї–Є–љ–µ–≤—А–Є—В–Є—З–µ—Б–Ї–Є–µ –љ–∞—А—Г—И–µ–љ–Є—П¬†вАУ –Њ–љ–µ–Љ–µ–љ–Є–µ –Є¬†–њ–∞—А–µ—Б—В–µ–Ј–Є–Є –≤¬†–Ї–Є—Б—В—П—Е –Є¬†—Б—В–Њ–њ–∞—Е.
–Э–∞–њ—А–∞–≤–Є—В–µ–ї—М–љ—Л–Љ–Є –і–Є–∞–≥–љ–Њ–Ј–∞–Љ–Є, —Б¬†–Ї–Њ—В–Њ—А—Л–Љ–Є –њ–∞—Ж–Є–µ–љ—В—Л —Б¬†–Ґ–Ґ–†-–°–Р–Я –Њ–±—А–∞—Й–∞–ї–Є—Б—М –≤¬†–§–У–С–Э–£ ¬Ђ–Э–∞—Г—З–љ—Л–є —Ж–µ–љ—В—А –љ–µ–≤—А–Њ–ї–Њ–≥–Є–Є¬ї, –±—Л–ї–Є –Я–Э–Я –љ–µ—П—Б–љ–Њ–≥–Њ –≥–µ–љ–µ–Ј–∞ –Є–ї–Є –Є–і–Є–Њ–њ–∞—В–Є—З–µ—Б–Ї–∞—П –Я–Э–Я (25 (71%) –±–Њ–ї—М–љ—Л—Е), –•–Т–Ф–Я (5¬†(14%)), —А–∞–і–Є–Ї—Г–ї–Њ–њ–∞—В–Є—П (2 (6%)), –љ–µ–є—А–Њ–і–µ–≥–µ–љ–µ—А–∞—В–Є–≤–љ–Њ–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ –љ–µ—А–≤–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л (2 (6%)) –Є¬†–±–Њ–ї–µ–Ј–љ—М –і–≤–Є–≥–∞—В–µ–ї—М–љ–Њ–≥–Њ –љ–µ–є—А–Њ–љ–∞ (1 (3%)).
–•–∞—А–∞–Ї—В–µ—А–Є—Б—В–Є–Ї–∞ –ґ–∞–ї–Њ–±, —А–µ–Ј—Г–ї—М—В–∞—В—Л –Њ–±—Й–µ–≥–Њ –Є¬†–љ–µ–≤—А–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–≥–Њ –Њ—Б–Љ–Њ—В—А–∞
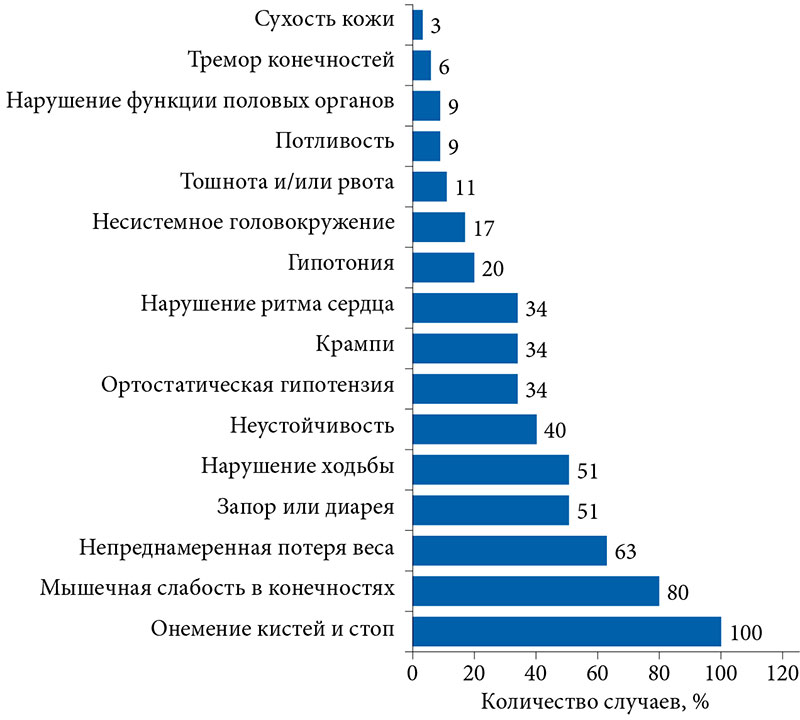
–Э–∞¬†–Љ–Њ–Љ–µ–љ—В –Њ–±—А–∞—Й–µ–љ–Є—П –≤¬†–§–У–С–Э–£ ¬Ђ–Э–∞—Г—З–љ—Л–є —Ж–µ–љ—В—А –љ–µ–≤—А–Њ–ї–Њ–≥–Є–Є¬ї –Њ—Б–љ–Њ–≤–љ—Л–Љ–Є –ґ–∞–ї–Њ–±–∞–Љ–Є –±—Л–ї–Є –Њ–љ–µ–Љ–µ–љ–Є–µ –Ї–Є—Б—В–µ–є –Є¬†—Б—В–Њ–њ (35 (100%) –њ–∞—Ж–Є–µ–љ—В–Њ–≤), –Љ—Л—И–µ—З–љ–∞—П —Б–ї–∞–±–Њ—Б—В—М –≤¬†–Ї–Њ–љ–µ—З–љ–Њ—Б—В—П—Е (28 (80%)), –љ–µ–њ—А–µ–і–љ–∞–Љ–µ—А–µ–љ–љ–Њ–µ —Б–љ–Є–ґ–µ–љ–Є–µ –≤–µ—Б–∞ (22 (63%)), –Ј–∞–њ–Њ—А –Є–ї–Є –і–Є–∞—А–µ—П (18¬†(51%)) –Є¬†–љ–∞—А—Г—И–µ–љ–Є–µ —Е–Њ–і—М–±—Л (18 (51%)) (—А–Є—Б—Г–љ–Њ–Ї).
–Ь–µ–і–Є–∞–љ–∞, –∞¬†—В–∞–Ї–ґ–µ –≤–µ—А—Е–љ–Є–є –Є¬†–љ–Є–ґ–љ–Є–є –Ї–≤–∞—А—В–Є–ї–Є (Q1; Q4) –Є–љ–і–µ–Ї—Б–∞ –Љ–∞—Б—Б—Л —В–µ–ї–∞ –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б–Њ—Б—В–∞–≤–Є–ї–Є 21,15 –Ї–≥/–Љ2 [28,4; 17,4].
–Я—А–Є –љ–µ–≤—А–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–Љ –Њ—Б–Љ–Њ—В—А–µ —Г¬†–≤–Ї–ї—О—З–µ–љ–љ—Л—Е –≤¬†–Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –Њ–±–љ–∞—А—Г–ґ–µ–љ—Л –њ–Њ–ї–Є–љ–µ–≤—А–Є—В–Є—З–µ—Б¬≠–Ї–Є–µ –љ–∞—А—Г—И–µ–љ–Є—П —А–∞–Ј–љ–Њ–є —Б—В–µ–њ–µ–љ–Є –≤—Л—А–∞–ґ–µ–љ–љ–Њ—Б—В–Є. –Ь–µ–і–Є–∞–љ–∞, –∞¬†—В–∞–Ї–ґ–µ –≤–µ—А—Е–љ–Є–є –Є¬†–љ–Є–ґ–љ–Є–є –Ї–≤–∞—А—В–Є–ї–Є (Q1; Q4) –Љ—Л—И–µ—З–љ–Њ–є —Б–Є–ї—Л, –Њ—Ж–µ–љ–Є–≤–∞–µ–Љ–Њ–є –њ–Њ¬†MRCss, –≤¬†–њ—А–Њ–Ї—Б–Є–Љ–∞–ї—М–љ—Л—Е –Њ—В–і–µ–ї–∞—Е —А—Г–Ї —Б–Њ—Б—В–∞–≤–Є–ї–Є 3,75 [4,00; 3,50] –±–∞–ї–ї–∞, –≤¬†–і–Є—Б—В–∞–ї—М¬≠–љ—Л—Е –Њ—В–і–µ–ї–∞—Е —А—Г–Ї (–Ї–Є—Б—В—П—Е)¬†вАУ 3,5 [4,0; 2,0] –±–∞–ї–ї–∞, –≤¬†–њ—А–Њ–Ї—Б–Є–Љ–∞–ї—М–љ—Л—Е –Њ—В–і–µ–ї–∞—Е –љ–Њ–≥¬†вАУ 3,5 [4,0; 3,0] –±–∞–ї–ї–∞, –≤¬†–і–Є—Б—В–∞–ї—М–љ—Л—Е –Њ—В–і–µ–ї–∞—Е –љ–Њ–≥ (—Б—В–Њ–њ–∞—Е)¬†вАУ 2,0 [4,0; 0,0] –±–∞–ї–ї–∞.
–Я–µ—А–≤–Њ–є –Є¬†–≤—В–Њ—А–Њ–є —Б—В–∞–і–Є—П–Љ –Ґ–Ґ–†-–°–Р–Я —Б–Њ–Њ—В–≤–µ—В—Б—В–≤–Њ–≤–∞–ї–Є –њ–Њ¬†14 –њ–∞—Ж–Є–µ–љ—В–Њ–≤, —В—А–µ—В—М–µ–є —Б—В–∞–і–Є–Є¬†вАУ 7.
–•–∞—А–∞–Ї—В–µ—А–Є—Б—В–Є–Ї–∞ —Б–Є—Б—В–µ–Љ–љ—Л—Е –Њ–±—Й–µ—Б–Њ–Љ–∞—В–Є—З–µ—Б–Ї–Є—Е –љ–∞—А—Г—И–µ–љ–Є–є
–Я–∞—В–Њ–ї–Њ–≥–Є—П –ґ–µ–ї—Г–і–Њ—З–љ–Њ-–Ї–Є—И–µ—З–љ–Њ–≥–Њ —В—А–∞–Ї—В–∞ –≤—Б—В—А–µ—З–∞–ї–∞—Б—М –±–Њ–ї–µ–µ —З–µ–Љ –≤¬†–њ–Њ–ї–Њ–≤–Є–љ–µ (22 (63%)) —Б–ї—Г—З–∞–µ–≤, –≤—Л–Ј—Л–≤–∞—П –љ–µ–њ—А–µ–і–љ–∞–Љ–µ—А–µ–љ–љ—Г—О –њ–Њ—В–µ—А—О –≤–µ—Б–∞ (22 (63%)), –і–Є–∞—А–µ—О –Є¬†–Ј–∞–њ–Њ—А—Л (18 (51%)), —В–Њ—И–љ–Њ—В—Г –Є¬†—А–≤–Њ—В—Г (4 (11%)). –Ф–Њ–њ–Њ–ї–љ–Є—В–µ–ї—М–љ–Њ–µ –Њ–±—Й–µ—Б–Њ–Љ–∞—В–Є—З–µ—Б–Ї–Њ–µ –Њ–±—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –њ–Њ–Ї–∞–Ј–∞–ї–Њ, —З—В–Њ —Б–ї–µ–і—Г—О—Й–Є–Љ –њ–Њ¬†—З–∞—Б—В–Њ—В–µ —Б–Є—Б—В–µ–Љ–љ—Л–Љ –њ—А–Њ—П–≤–ї–µ–љ–Є–µ–Љ –±–Њ–ї–µ–Ј–љ–Є –±—Л–ї–∞ –њ–∞—В–Њ–ї–Њ–≥–Є—П —Б–µ—А–і–µ—З–љ–Њ-—Б–Њ—Б—Г–і–Є—Б—В–Њ–є —Б–Є—Б—В–µ–Љ—Л (12 (34%) —Б–ї—Г—З–∞–µ–≤), –Ї–Њ—В–Њ—А–∞—П –≤–Ї–ї—О—З–∞–ї–∞ –љ–∞—А—Г—И–µ–љ–Є–µ —А–Є—В–Љ–∞ —Б–µ—А–і—Ж–∞ (6 (17%)), –Ї–∞—А–і–Є–Њ–Љ–Є–Њ–њ–∞—В–Є—О (—В–Њ–ї—Й–Є–љ–∞ –Љ–µ–ґ–ґ–µ–ї—Г–і–Њ—З–Ї–Њ–≤–Њ–є –њ–µ—А–µ–≥–Њ—А–Њ–і–Ї–Є/–Ј–∞–і–љ–µ–є —Б—В–µ–љ–Ї–Є –ї–µ–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞ > 11 –Љ–Љ) (5¬†(14%)), —В–∞—Е–Є–Ї–∞—А–і–Є—О (4 (11%)) –Є¬†–±–ї–Њ–Ї–∞–і—Г –њ—А–Њ–≤–Њ–і—П—Й–µ–є —Б–Є—Б—В–µ–Љ—Л —Б–µ—А–і—Ж–∞ (2 (6%)). –†–∞–і–Є–Њ—З–∞—Б—В–Њ—В–љ–∞—П –Ї–∞—В–µ—В–µ—А–љ–∞—П –∞–±–ї—П—Ж–Є—П –њ–Њ¬†–њ–Њ–≤–Њ–і—Г –њ–∞—А–Њ–Ї—Б–Є–Ј–Љ–∞–ї—М–љ–Њ–є —Д–Њ—А–Љ—Л —Д–Є–±—А–Є–ї–ї—П—Ж–Є–Є –њ—А–µ–і—Б–µ—А–і–Є–є –≤¬†–∞–љ–∞–Љ–љ–µ–Ј–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –±—Л–ї–∞ –њ—А–Њ–≤–µ–і–µ–љ–∞ 2 (6%) –њ–∞—Ж–Є–µ–љ—В–∞–Љ.
–†–µ–ґ–µ —Г¬†–±–Њ–ї—М–љ—Л—Е –Ґ–Ґ–†-–°–Р–Я –≤—Л—П–≤–ї—П–ї–Њ—Б—М –њ–Њ–Љ—Г—В–љ–µ–љ–Є–µ —Б—В–µ–Ї–ї–Њ–≤–Є–і–љ–Њ–≥–Њ —В–µ–ї–∞ –Є¬†—Б–љ–Є–ґ–µ–љ–Є–µ –Ј—А–µ–љ–Є—П (5 (14%)), –∞¬†—В–∞–Ї–ґ–µ –њ–∞—В–Њ–ї–Њ–≥–Є—П –њ–Њ—З–µ–Ї (4 (11%)), –Ї–Њ—В–Њ—А–∞—П –≤–Ї–ї—О—З–∞–ї–∞ –Љ–Є–Ї—А–Њ–њ—А–Њ—В–µ–Є–љ—Г—А–Є—О –і–Њ 0,06 –≥/–ї (2 (6%)) –Є¬†—Е—А–Њ–љ–Є—З–µ—Б¬≠–Ї—Г—О –±–Њ–ї–µ–Ј–љ—М –њ–Њ—З–µ–Ї (4 (11%)).
–Р–љ–∞–ї–Є–Ј —А–µ–Ј—Г–ї—М—В–∞—В–Њ–≤ –ї–∞–±–Њ—А–∞—В–Њ—А–љ–Њ–≥–Њ –Є¬†–Є–љ—Б—В—А—Г–Љ–µ–љ—В–∞–ї—М–љ–Њ–≥–Њ –Њ–±—Б–ї–µ–і–Њ–≤–∞–љ–Є—П
–£¬†11 (31%) –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –±—Л–ї –≤–Ј—П—В –∞–љ–∞–ї–Є–Ј –Ї—А–Њ–≤–Є –љ–∞¬†–Љ–Њ–Ј–≥–Њ–≤–Њ–є –љ–∞—В—А–Є–є—Г—А–µ—В–Є—З–µ—Б–Ї–Є–є –≥–Њ—А–Љ–Њ–љ (NT-proBNP). –Я–Њ–≤—Л—И–µ–љ–љ—Л–є —Г—А–Њ–≤–µ–љ—М NT-proBNP (–±–Њ–ї–µ–µ 125 –њ–≥/–Љ–ї) –±—Л–ї –≤—Л—П–≤–ї–µ–љ —Г¬†6 (55%) –њ–∞—Ж–Є–µ–љ—В–Њ–≤. –£—А–Њ–≤–µ–љ—М —В—А–Њ–њ–Њ–љ–Є–љ–∞ –Њ–њ—А–µ–і–µ–ї–µ–љ —Г¬†4 (11%) –њ–∞—Ж–Є–µ–љ—В–Њ–≤, –Є¬†–ї–Є—И—М —Г¬†1 (25%) –Є–Ј¬†–љ–Є—Е –Њ—В–Љ–µ—З–∞–ї–Њ—Б—М –µ–≥–Њ –њ–Њ–≤—Л—И–µ–љ–Є–µ –і–Њ 42 –љ–≥/–Љ–ї.
–Ф–Њ –Њ–±—А–∞—Й–µ–љ–Є—П –≤¬†–§–У–С–Э–£ ¬Ђ–Э–∞—Г—З–љ—Л–є —Ж–µ–љ—В—А –љ–µ–≤—А–Њ–ї–Њ–≥–Є–Є¬ї 4 (11%) –њ–∞—Ж–Є–µ–љ—В–∞–Љ –±—Л–ї–∞ –њ—А–Њ–≤–µ–і–µ–љ–∞ –ї—О–Љ–±–∞–ї—М–љ–∞—П –њ—Г–љ–Ї—Ж–Є—П —Б¬†–њ–Њ—Б–ї–µ–і—Г—О—Й–Є–Љ –Њ–±—Й–Є–Љ –∞–љ–∞–ї–Є–Ј–Њ–Љ –ї–Є–Ї–≤–Њ—А–∞. –Т¬†2 (50%) —Б–ї—Г—З–∞—П—Е –≤—Л—П–≤–ї–µ–љ–Њ –њ–Њ–≤—Л—И–µ–љ–Є–µ —Г—А–Њ–≤–љ—П –±–µ–ї–Ї–∞ –±–Њ–ї–µ–µ 0,45 –≥/–ї.
–Я—А–Є –њ—А–Њ–≤–µ–і–µ–љ–Є–Є –≠–Э–Ь–У –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Є—Е –љ–µ—А–≤–Њ–≤ —Г¬†31¬†(89%) –њ–∞—Ж–Є–µ–љ—В–∞ —Б¬†–Ґ–Ґ–†-–°–Р–Я —А–µ–≥–Є—Б—В—А–Є—А–Њ–≤–∞–ї–Є—Б—М –љ–µ–є—А–Њ—Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –њ—А–Є–Ј–љ–∞–Ї–Є –≥–µ–љ–µ—А–∞–ї–Є–Ј–Њ–≤–∞–љ–љ–Њ–≥–Њ —Б–Є–Љ–Љ–µ—В—А–Є—З–љ–Њ–≥–Њ —Б–µ–љ—Б–Њ–Љ–Њ—В–Њ—А–љ–Њ–≥–Њ –љ–µ–≤—А–∞–ї—М–љ–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П, –±–Њ–ї–µ–µ –≤—Л—А–∞–ґ–µ–љ–љ–Њ–≥–Њ –≤¬†–љ–Є–ґ–љ–Є—Е –Ї–Њ–љ–µ—З–љ–Њ—Б—В—П—Е. –Т¬†–њ–Њ–і–∞–≤–ї—П—О—Й–µ–Љ –±–Њ–ї—М—И–Є–љ—Б—В–≤–µ (27 (87%)) —Б–ї—Г—З–∞–µ–≤ —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ –∞–Ї—Б–Њ–љ–∞–ї—М–љ—Л–є —Е–∞—А–∞–Ї—В–µ—А –љ–µ–≤—А–∞–ї—М–љ–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П, –≤¬†–Љ–µ–љ—М—И–µ–Љ –Ї–Њ–ї–Є—З–µ—Б—В–≤–µ (4 (13%) —Б–ї—Г—З–∞–µ–≤¬†вАУ –і–µ–Љ–Є–µ–ї–Є–љ–Є–Ј–Є—А—Г—Й–Є–є —Е–∞—А–∞–Ї—В–µ—А –љ–µ–≤—А–∞–ї—М–љ–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П.
–†–µ–ґ–µ (8 (23%)) –њ—А–Є –≠–Э–Ь–У –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Є—Е –љ–µ—А–≤–Њ–≤ –Њ—В–Љ–µ—З–∞–ї–Є—Б—М –љ–µ–є—А–Њ—Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –њ—А–Є–Ј–љ–∞–Ї–Є –Ї–Њ–Љ–њ—А–µ—Б—Б–Є–Њ–љ–љ—Л—Е –Љ–Њ–љ–Њ–љ–µ–є—А–Њ–њ–∞—В–Є–є. –Ґ–∞–Ї, –ї–Њ–Ї–∞–ї—М–љ–Њ–µ –љ–∞—А—Г—И–µ–љ–Є–µ –њ—А–Њ–≤–µ–і–µ–љ–Є—П –≤–Њ–Ј–±—Г–ґ–і–µ–љ–Є—П –њ–Њ¬†—Б—А–µ–і–Є–љ–љ–Њ–Љ—Г –љ–µ—А–≤—Г –љ–∞¬†—Г—А–Њ–≤–љ–µ –Ї–∞—А–њ–∞–ї—М–љ–Њ–≥–Њ –Ї–∞–љ–∞–ї–∞ –Њ–±–љ–∞—А—Г–ґ–µ–љ–Њ —Г¬†8¬†(23%) –њ–∞—Ж–Є–µ–љ—В–Њ–≤, —Б–Њ—З–µ—В–∞–љ–Є–µ –љ–∞—А—Г—И–µ–љ–Є—П –њ—А–Њ–≤–µ–і–µ–љ–Є—П –≤–Њ–Ј–±—Г–ґ–і–µ–љ–Є—П –њ–Њ¬†—Б—А–µ–і–Є–љ–љ–Њ–Љ—Г –Є¬†–ї–Њ–Ї—В–µ–≤–Њ–Љ—Г –љ–µ—А–≤–∞–Љ –љ–∞¬†—Г—А–Њ–≤–љ–µ —В—Г–љ–љ–µ–ї–µ–є (–Ї–∞—А–њ–∞–ї—М–љ—Л–є –Є¬†–Ї—Г–±–Є—В–∞–ї—М–љ—Л–є –Ї–∞–љ–∞–ї—Л —Б–Њ–Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ)¬†вАУ —Г¬†3 (8,5%).
–°–Њ—З–µ—В–∞–љ–Є–µ –љ–µ–є—А–Њ—Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е –њ—А–Є–Ј–љ–∞–Ї–Њ–≤ –≥–µ–љ–µ—А–∞–ї–Є–Ј–Њ–≤–∞–љ–љ–Њ–≥–Њ –њ–Њ–ї–Є–љ–µ–≤—А–Є—В–Є—З–µ—Б–Ї–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П —Б¬†–ї–Њ–Ї–∞–ї—М–љ—Л–Љ –љ–∞—А—Г—И–µ–љ–Є–µ–Љ –њ—А–Њ–≤–µ–і–µ–љ–Є—П –≤–Њ–Ј–±—Г–ґ–і–µ–љ–Є—П –њ–Њ¬†—Б—А–µ–і–Є–љ–љ–Њ–Љ—Г –љ–µ—А–≤—Г –љ–∞¬†—Г—А–Њ–≤–љ–µ –Ї–∞—А–њ–∞–ї—М–љ–Њ–≥–Њ –Ї–∞–љ–∞–ї–∞ –≤—Л—П–≤–ї–µ–љ–Њ —Г¬†4 (11%) –њ–∞—Ж–Є–µ–љ—В–Њ–≤.
–£–ї—М—В—А–∞–Ј–≤—Г–Ї–Њ–≤–Њ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Є—Е –љ–µ—А¬≠–≤–Њ–≤, –≤—Л–њ–Њ–ї–љ–µ–љ–љ–Њ–µ –њ–Њ¬†–њ—А–Њ—В–Њ–Ї–Њ–ї—Г UPSS, –њ—А–Њ–≤–µ–і–µ–љ–Њ 9¬†–Є–Ј¬†35 –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–Ґ–Ґ–†-–°–Р–Я. –£¬†3 (33%) –Є–Ј¬†–љ–Є—Е —Б—Г–Љ–Љ–∞—А–љ—Л–є –±–∞–ї–ї —Б–Њ—Б—В–∞–≤–Є–ї –і–µ–≤—П—В—М –Є¬†–±–Њ–ї–µ–µ, —З—В–Њ —Б–Њ–Њ—В–≤–µ—В—Б—В–≤—Г–µ—В –Є–Ј–Љ–µ–љ–µ–љ–Є—П–Љ, —Е–∞—А–∞–Ї—В–µ—А–љ—Л–Љ –і–ї—П –•–Т–Ф–Я. –£¬†6¬†(67%) –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –±—Л–ї–Є –Љ—П–≥–Ї–Є–Љ–Є –Є–ї–Є –≤–Њ–≤—Б–µ –Њ—В—Б—Г—В—Б—В–≤–Њ–≤–∞–ї–Є. –Ь–µ–і–Є–∞–љ–∞, –∞¬†—В–∞–Ї–ґ–µ –≤–µ—А—Е–љ–Є–є –Є¬†–љ–Є–ґ–љ–Є–є –Ї–≤–∞—А—В–Є–ї–Є (Q1; Q4) –њ–Њ¬†–њ—А–Њ—В–Њ–Ї–Њ–ї—Г UPSS —Б–Њ—Б—В–∞–≤–Є–ї–Є 4 [11; 0] –±–∞–ї–ї–∞.
–Ш–Ј¬†7 (20%) –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–Ґ–Ґ–†-–°–Р–Я, –Ї–Њ—В–Њ—А—Л–Љ –±—Л–ї–Њ –њ—А–Њ–≤–µ–і–µ–љ–Њ –Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –Є–Ї—А–Њ–љ–Њ–ґ–љ–Њ–≥–Њ –љ–µ—А–≤–∞ –Є¬†–њ–Њ–і–Ї–Њ–ґ–љ–Њ-–ґ–Є—А–Њ–≤–Њ–є –Ї–ї–µ—В—З–∞—В–Ї–Є, —В–Њ–ї—М–Ї–Њ —Г¬†4 (57%) –≤—Л—П–≤–ї–µ–љ—Л –Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –њ—А–Є–Ј–љ–∞–Ї–Є –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞.
–Ю–±—Б—Г–ґ–і–µ–љ–Є–µ
–Я—А–Є –њ—А–Њ–≤–µ–і–µ–љ–Є–Є –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ–Њ-–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Њ–≥–Њ —Б–Ї—А–Є–љ–Є–љ–≥–∞ 750 —А–Њ—Б—Б–Є–є—Б–Ї–Є–Љ –њ–∞—Ж–Є–µ–љ—В–∞–Љ —Б¬†–њ—А–Є–Љ–µ–љ–µ–љ–Є–µ–Љ –∞–ї–≥–Њ—А–Є—В–Љ–∞ –њ–Њ–Є—Б–Ї–∞ –њ–Њ¬†–Ї–ї—О—З–µ–≤—Л–Љ –і–Є–∞–≥–љ–Њ–Ј–∞–Љ –Є¬†–љ–∞–ї–Є—З–Є—О –і–Њ–њ–Њ–ї–љ–Є—В–µ–ї—М–љ—Л—Е ¬Ђ–Ї—А–∞—Б–љ—Л—Е —Д–ї–∞–ґ–Ї–Њ–≤¬ї –і–Є–∞–≥–љ–Њ–Ј –Ґ–Ґ–†-–°–Р–Я –±—Л–ї –њ–Њ–і—В–≤–µ—А–ґ–і–µ–љ —Г¬†35 (5%). –Ф–∞–љ–љ—Л–µ –Ј–∞—А—Г–±–µ–ґ–љ—Л—Е –Ї–Њ–ї–ї–µ–≥ –њ–Њ¬†–≤—Л—П–≤–ї—П–µ–Љ–Њ—Б—В–Є –Ґ–Ґ–†-–°–Р–Я –≤–∞—А—М–Є—А—Г—О—В—Б—П. –Ґ–∞–Ї, –≤¬†–Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є G. Fargeot –Є¬†—Б–Њ–∞–≤—В. (2024 –≥.), –≤¬†–Ї–Њ—В–Њ—А–Њ–Љ –њ—А–Є–љ—П–ї–Є —Г—З–∞—Б—В–Є–µ 553 –њ–∞—Ж–Є–µ–љ—В–∞, –њ–∞—В–Њ–≥–µ–љ–љ—Л–µ –Љ—Г—В–∞—Ж–Є–Є –≥–µ–љ–∞ TTR –±—Л–ї–Є –Њ–±–љ–∞—А—Г–ґ–µ–љ—Л —Г¬†15 (2,7%) [11]. –Т¬†–Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є V. Di Stefano –Є¬†—Б–Њ–∞–≤—В. (2023 –≥.) –Є–Ј¬†145¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ –њ–Њ–і—В–≤–µ—А–і–Є–ї–Њ—Б—М —Г¬†10% [12].
–Э–∞¬†–Њ—Б–љ–Њ–≤–∞–љ–Є–Є —А–µ–Ј—Г–ї—М—В–∞—В–Њ–≤ –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ–Њ-–≥–µ–љ–µ—В–Є—З–µ—Б¬≠–Ї–Њ–≥–Њ –Њ–±—Б–ї–µ–і–Њ–≤–∞–љ–Є—П, –њ—А–Њ–≤–µ–і–µ–љ–љ–Њ–≥–Њ –љ–∞¬†–±–∞–Ј–µ –§–У–С–Э–£ ¬Ђ–Э–∞—Г—З–љ—Л–є —Ж–µ–љ—В—А –љ–µ–≤—А–Њ–ї–Њ–≥–Є–Є¬ї, –±—Л–ї–∞ —Б—Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–∞ –±–∞–Ј–∞ –і–∞–љ–љ—Л—Е —Б–Є–Љ–њ—В–Њ–Љ–љ—Л—Е (n = 35) –Є¬†–±–µ—Б—Б–Є–Љ–њ—В–Њ–Љ–љ—Л—Е (n = 13) –љ–Њ—Б–Є—В–µ–ї–µ–є –Љ—Г—В–∞—Ж–Є–є –≥–µ–љ–∞ TTR. –Ґ–∞–Ї–Њ–є –њ–Њ–і—Е–Њ–і –≤–∞–ґ–µ–љ –і–ї—П –Ї–Њ–Љ–њ–ї–µ–Ї—Б–љ–Њ–≥–Њ –∞–љ–∞–ї–Є–Ј–∞ –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†—А–µ–і–Ї–Є–Љ–Є (–Њ—А—Д–∞–љ–љ—Л–Љ–Є) –±–Њ–ї–µ–Ј–љ—П–Љ–Є, –∞¬†—В–∞–Ї–ґ–µ –і–ї—П —Б—А–∞–≤–љ–µ–љ–Є—П –і–∞–љ–љ—Л—Е –њ–Њ¬†–†–Њ—Б—Б–Є–Є —Б¬†–і–∞–љ–љ—Л–Љ–Є –≤¬†–і—А—Г–≥–Є—Е —Б—В—А–∞–љ–∞—Е. –Э–∞–њ—А–Є–Љ–µ—А, –≤¬†–С—А–∞–Ј–Є–ї–Є–Є —Б–Њ–±—А–∞–љ–∞ –Ї–Њ–≥–Њ—А—В–∞ —Б¬†—А–∞—Б—Б–Љ–∞—В—А–Є–≤–∞–µ–Љ—Л–Љ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ–Љ, –Ї–Њ—В–Њ—А–∞—П –≤–Ї–ї—О—З–∞–µ—В 108 –њ–∞—Ж–Є–µ–љ—В–Њ–≤ [13]. –°–∞–Љ—Л–Љ –Ї—А—Г–њ–љ—Л–Љ —Б—З–Є—В–∞–µ—В—Б—П –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ THAOS, –Ї–Њ—В–Њ—А–Њ–µ –≤–Ї–ї—О—З–Є–ї–Њ –±–Њ–ї–µ–µ 6000 –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†—В—А–∞–љ—Б—В–Є—А–µ—В–Є–љ–Њ–≤—Л–Љ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–Њ–Љ –Є¬†–Њ–±—К–µ–і–Є–љ–Є–ї–Њ –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–Ґ–Ґ–†-–°–Р–Я, —В—А–∞–љ—Б—В–Є—А–µ—В–Є–љ–Њ–≤–Њ–є –∞–Љ–Є–ї–Њ–Є–і–љ–Њ–є –Ї–∞—А–і–Є–Њ–Љ–Є–Њ–њ–∞—В–Є–µ–є, –∞¬†—В–∞–Ї–ґ–µ —Б¬†–і–Є–Ї–Є–Љ —В–Є–њ–Њ–Љ —В—А–∞–љ—Б—В–Є—А–µ—В–Є–љ–Њ–≤–Њ–≥–Њ –∞–Љ–Є–ї–Њ–Є–і–Њ–Ј–∞ [14].
–Я—А–Є –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–Љ –Њ–±—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є –љ–∞—И–µ–є –≤—Л–±–Њ—А–Ї–Є –≤—Л—П–≤–ї–µ–љ—Л –љ–∞—А—Г—И–µ–љ–Є—П, —Б–Њ–Њ—В–≤–µ—В—Б—В–≤—Г—О—Й–Є–µ –Ї–ї–∞—Б—Б–Є—З–µ—Б–Ї–Њ–Љ—Г –њ–Њ—А—В—А–µ—В—Г –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П [15, 16]. –°–ї–µ–і—Г–µ—В –Њ—В–Љ–µ—В–Є—В—М, —З—В–Њ –≤–µ–≥–µ—В–∞—В–Є–≤–љ—Л–µ –љ–∞—А—Г—И–µ–љ–Є—П, —Б–Њ–њ—Г—В—Б—В–≤—Г—О—Й–Є–µ –Я–Э–Я, –Њ–±–љ–∞—А—Г–ґ–µ–љ—Л –≤¬†34% —Б–ї—Г—З–∞–µ–≤, –љ–µ–њ—А–µ–і–љ–∞–Љ–µ—А–µ–љ–љ–Њ–µ —Б–љ–Є–ґ–µ–љ–Є–µ –≤–µ—Б–∞¬†вАУ –≤¬†63%, –њ—А–Є–Ј–љ–∞–Ї–Є —Б–Є—Б—В–µ–Љ–љ–Њ—Б¬≠—В–Є¬†вАУ –≤¬†30%¬†—Б–ї—Г—З–∞–µ–≤, —З—В–Њ –±—Л–ї–Њ —Б–Њ–њ–Њ—Б—В–∞–≤–Є–Љ–Њ —Б¬†–і–∞–љ–љ—Л–Љ–Є –ї–Є—В–µ—А–∞—В—Г—А—Л. –Э–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В—Л–Љ–Є —Н–Ї—Б—В—А–∞–љ–µ–≤—А–∞–ї—М–љ—Л–Љ–Є –љ–∞—А—Г—И–µ–љ–Є—П–Љ–Є –Њ–Ї–∞–Ј–∞–ї–Є—Б—М –ґ–µ–ї—Г–і–Њ—З–љ–Њ-–Ї–Є—И–µ—З–љ—Л–µ —А–∞—Б—Б—В—А–Њ–є—Б—В–≤–∞ (63%) –Є¬†—Б–µ—А–і–µ—З–љ–Њ-—Б–Њ—Б—Г–і–Є—Б—В–∞—П –њ–∞—В–Њ–ї–Њ–≥–Є—П (34%), —З—В–Њ —В–∞–Ї–ґ–µ —Б–Њ–Њ—В–≤–µ—В—Б—В–≤—Г–µ—В –і–∞–љ–љ—Л–Љ –ї–Є—В–µ—А–∞—В—Г—А—Л¬†[12].
–Э–µ—Б–Љ–Њ—В—А—П –љ–∞¬†—В–Њ —З—В–Њ –њ–Њ—А–∞–ґ–µ–љ–Є–µ –≥–ї–∞–Ј –≤—Л—П–≤–ї—П–µ—В—Б—П —Г¬†–±–Њ–ї—М—И–Є–љ—Б—В–≤–∞ –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–Ґ–Ґ–†-–°–Р–Я (—Б–Є–љ–і—А–Њ–Љ —Б—Г—Е–Њ–≥–Њ –≥–ї–∞–Ј–∞¬†вАУ —Г¬†70%, –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П –Ј—А–∞—З–Ї–Њ–≤ –≤¬†–≤–Є–і–µ –Ј—Г–±—З–∞—В–Њ–є —А–∞–і—Г–ґ–љ–Њ–є –Њ–±–Њ–ї–Њ—З–Ї–Є¬†вАУ —Г¬†28%, –≥–ї–∞—Г–Ї–Њ–Љ–∞¬†вАУ —Г¬†20%, –њ–Њ–Љ—Г—В–љ–µ–љ–Є–µ —Б—В–µ–Ї–ї–Њ–≤–Є–і–љ–Њ–≥–Њ —В–µ–ї–∞¬†вАУ —Г¬†17%, –∞–љ–Њ–Љ–∞–ї—М–љ—Л–µ —Б–Њ—Б—Г–і—Л –Ї–Њ–љ—К—О–љ–Ї—В–Є–≤—Л¬†вАУ —Г¬†14%) [17], –≤¬†–љ–∞—И–µ–є –Ї–Њ–≥–Њ—А—В–µ –њ–Њ—А–∞–ґ–µ–љ–Є–µ –≥–ї–∞–Ј –±—Л–ї–Њ –Њ—В–Љ–µ—З–µ–љ–Њ –ї–Є—И—М —Г¬†14%. –Я–Њ–і–Њ–±–љ–∞—П –Ї–∞—А—В–Є–љ–∞ –љ–∞–±–ї—О–і–∞–ї–∞—Б—М –Є¬†–њ—А–Є –∞–љ–∞–ї–Є–Ј–µ —З–∞—Б—В–Њ—В—Л –≤—Б—В—А–µ—З–∞–µ–Љ–Њ—Б—В–Є –њ–∞—В–Њ–ї–Њ–≥–Є–Є –њ–Њ—З–µ–Ї. –Ґ–∞–Ї, –њ–∞—В–Њ–ї–Њ–≥–Є—П –њ–Њ—З–µ–Ї –≤–µ—А–Є—Д–Є—Ж–Є—А—Г–µ—В—Б—П –њ—А–Є–Љ–µ—А–љ–Њ —Г¬†30% –њ–∞—Ж–Є–µ–љ—В–Њ–≤ [17]. –Т¬†–љ–∞—И–µ–є –≤—Л–±–Њ—А–Ї–µ –≤–Њ–≤–ї–µ—З–µ–љ–Є–µ –њ–Њ—З–µ–Ї –≤¬†–њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–є –њ—А–Њ—Ж–µ—Б—Б –±—Л–ї–Њ –Ј–∞—Д–Є–Ї—Б–Є—А–Њ–≤–∞–љ–Њ –ї–Є—И—М –≤¬†11% —Б–ї—Г—З–∞–µ–≤.
–Ю—Б–Њ–±–Њ–≥–Њ –≤–љ–Є–Љ–∞–љ–Є—П –Ј–∞—Б–ї—Г–ґ–Є–≤–∞—О—В —А–µ–Ј—Г–ї—М—В–∞—В—Л –љ–µ–є—А–Њ—Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–≥–Њ –Њ–±—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Є—Е –љ–µ—А¬≠–≤–Њ–≤. –Т¬†4 (13%) —Б–ї—Г—З–∞—П—Е –Є–Ј 31 –њ—А–Є –њ—А–Њ–≤–µ–і–µ–љ–Є–Є –≠–Э–Ь–У –Њ–±–љ–∞—А—Г–ґ–µ–љ –і–µ–Љ–Є–µ–ї–Є–љ–Є–Ј–Є—А—Г—О—Й–Є–є —Е–∞—А–∞–Ї—В–µ—А –≥–µ–љ–µ—А–∞–ї–Є–Ј–Њ–≤–∞–љ–љ–Њ–≥–Њ –љ–µ–≤—А–∞–ї—М–љ–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П –Є¬†—А–∞–Ј–љ–∞—П —Б—В–µ–њ–µ–љ—М —Б–Њ–Њ—В–≤–µ—В—Б—В–≤–Є—П —Н–ї–µ–Ї—В—А–Њ–і–Є–∞–≥–љ–Њ—Б—В–Є—З–µ—Б–Ї–Є–Љ –Ї—А–Є—В–µ—А–Є—П–Љ –•–Т–Ф–Я, —З—В–Њ –≤¬†—А—П–і–µ —Б–ї—Г—З–∞–µ–≤ –њ—А–Є–≤–µ–ї–Њ –Ї¬†–љ–µ–≤–µ—А–љ–Њ–є –њ–Њ—Б—В–∞–љ–Њ–≤–Ї–µ –і–Є–∞–≥–љ–Њ–Ј–∞. –Я–Њ–ї—Г—З–µ–љ–љ—Л–µ –љ–∞–Љ–Є –і–∞–љ–љ—Л–µ —Б–Њ¬≠–њ–Њ—Б—В–∞–≤–Є–Љ—Л —Б¬†–і–∞–љ–љ—Л–Љ–Є –ї–Є—В–µ—А–∞—В—Г—А—Л, —Б–Њ–≥–ї–∞—Б–љ–Њ –Ї–Њ—В–Њ—А—Л–Љ –і–µ–Љ–Є–µ–ї–Є–љ–Є–Ј–Є—А—Г—О—Й–Є–є —Е–∞—А–∞–Ї—В–µ—А –њ–Њ—А–∞–ґ–µ–љ–Є—П –љ–µ—А–≤–Њ–≤ —А–µ–≥–Є—Б—В—А–Є—А–Њ–≤–∞–ї—Б—П —Г¬†15% –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–Ґ–Ґ–†-–°–Р–Я [18].
–£—Б—В–∞–љ–Њ–≤–ї–µ–љ–Њ, —З—В–Њ –њ—А–Є —Б–Њ–љ–Њ–≥—А–∞—Д–Є—З–µ—Б–Ї–Њ–Љ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Є—Е –љ–µ—А–≤–Њ–≤ —Г¬†18% –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–Ґ–Ґ–†-–°–Р–Я –Є–Љ–µ–ї–Њ –Љ–µ—Б—В–Њ —Г–≤–µ–ї–Є—З–µ–љ–Є–µ –њ–Њ–њ–µ—А–µ—З–љ–Њ–≥–Њ —Б–µ—З–µ–љ–Є—П —Б—В–≤–Њ–ї–Њ–≤ –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Є—Е –љ–µ—А–≤–Њ–≤ [19]. –Ь—Л –њ—А–Њ–≤–µ–ї–Є —Г–ї—М—В—А–∞–Ј–≤—Г–Ї–Њ–≤–Њ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Є—Е –љ–µ—А–≤–Њ–≤ –њ–Њ¬†–њ—А–Њ—В–Њ–Ї–Њ–ї—Г, –њ—А–µ–і–љ–∞–Ј–љ–∞—З–µ–љ–љ–Њ–Љ—Г –і–ї—П –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–Є –Є¬†–і–Є—Д—Д–µ—А–µ–љ—Ж–Є–∞–ї—М–љ–Њ–є –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–Є –•–Т–Ф–Я. –Т¬†–љ–∞—И–µ–є –≤—Л–±–Њ—А–Ї–µ –≤¬†3 (33%) —Б–ї—Г—З–∞—П—Е —Б—Г–Љ–Љ–∞—А–љ—Л–є –±–∞–ї–ї —Б–Њ—Б—В–∞–≤–Є–ї –і–µ–≤—П—В—М –Є¬†–±–Њ–ї–µ–µ, —З—В–Њ —Б–Њ–Њ—В–≤–µ—В—Б—В–≤—Г–µ—В –Є–Ј–Љ–µ–љ–µ–љ–Є—П–Љ, —Е–∞—А–∞–Ї—В–µ—А–љ—Л–Љ –і–ї—П –•–Т–Ф–Я. –Т¬†6 (67%) —Б–ї—Г—З–∞—П—Е –Є–Ј–Љ–µ–љ–µ–љ–Є—П –±—Л–ї–Є –Љ—П–≥–Ї–Є–Љ–Є –Є–ї–Є –≤–Њ–≤—Б–µ –Њ—В—Б—Г—В—Б—В–≤–Њ–≤–∞–ї–Є.
–Ь–µ–і–Є–∞–љ–∞, –∞¬†—В–∞–Ї–ґ–µ –≤–µ—А—Е–љ–Є–є –Є¬†–љ–Є–ґ–љ–Є–є –Ї–≤–∞—А—В–Є–ї–Є (Q1; Q4) –њ–Њ¬†–њ—А–Њ—В–Њ–Ї–Њ–ї—Г UPSS —Б–Њ—Б—В–∞–≤–Є–ї–Є 4 [11; 0] –±–∞–ї–ї–∞.
–Ш–Ј–≤–µ—Б—В–љ–Њ, —З—В–Њ –Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –њ–Њ–Ј–≤–Њ–ї—П–µ—В –њ–Њ–і—В–≤–µ—А–і–Є—В—М –і–Є–∞–≥–љ–Њ–Ј –Ґ–Ґ–†-–°–Р–Я –≤¬†86% —Б–ї—Г—З–∞–µ–≤. –Ю–і–љ–∞–Ї–Њ –љ–µ—А–∞–≤–љ–Њ–Љ–µ—А–љ–Њ–µ —А–∞—Б–њ—А–µ–і–µ–ї–µ–љ–Є–µ –Њ—В–ї–Њ–ґ–µ–љ–Є–є –∞–Љ–Є–ї–Њ–Є–і–∞ –Љ–Њ–ґ–µ—В –Њ–≥—А–∞–љ–Є—З–Є–≤–∞—В—М –і–Є–∞–≥–љ–Њ—Б—В–Є—З–µ—Б–Ї—Г—О —Ж–µ–љ–љ–Њ—Б—В—М –і–∞–љ–љ–Њ–≥–Њ –Љ–µ—В–Њ–і–∞, –њ–Њ—В–µ–љ—Ж–Є–∞–ї—М–љ–Њ —В—А–µ–±—Г—П –њ–Њ–≤—В–Њ—А–љ—Л—Е –±–Є–Њ–њ—Б–Є–є –љ–µ—А–≤–Њ–≤ –Є–ї–Є –і—А—Г–≥–Є—Е –њ–Њ—А–∞–ґ–µ–љ–љ—Л—Е –Њ—А–≥–∞–љ–Њ–≤ [2]. –Т¬†–љ–∞—И–µ–є –≤—Л–±–Њ—А–Ї–µ 7 (20%) –њ–∞—Ж–Є–µ–љ—В–∞–Љ —Б¬†–Ґ–Ґ–†-–°–Р–Я –±—Л–ї–Њ –њ—А–Њ–≤–µ–і–µ–љ–Њ –Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –Є–Ї—А–Њ–љ–Њ–ґ–љ–Њ–≥–Њ –љ–µ—А–≤–∞, –Ї–Њ—В–Њ—А–Њ–µ –≤—Л—П–≤–Є–ї–Њ —Е–∞—А–∞–Ї—В–µ—А–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –ї–Є—И—М —Г¬†4 (57%) –њ–∞—Ж–Є–µ–љ—В–Њ–≤.
–Ч–∞–Ї–ї—О—З–µ–љ–Є–µ
–Т¬†—Н–њ–Њ—Е—Г —Б–Њ–≤–µ—А—И–µ–љ—Б—В–≤–Њ–≤–∞–љ–Є—П —В–∞—А–≥–µ—В–љ–Њ–є –њ–∞—В–Њ–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Њ–є —В–µ—А–∞–њ–Є–Є –Ґ–Ґ–†-–°–Р–Я –∞–Ї—В—Г–∞–ї—М–љ–Њ—Б—В—М —Б–≤–Њ–µ–≤—А–µ–Љ–µ–љ–љ–Њ–≥–Њ –≤—Л—П–≤–ї–µ–љ–Є—П –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П —З—А–µ–Ј–≤—Л—З–∞–є–љ–Њ –≤—Л—Б–Њ–Ї–∞. –Я—А–Є –љ–∞–ї–Є—З–Є–Є ¬Ђ–Ї—А–∞—Б–љ—Л—Е —Д–ї–∞–ґ–Ї–Њ–≤¬ї —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–µ–є —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–є –њ–Њ–ї–Є–љ–µ–є—А–Њ–њ–∞—В–Є–µ–є –њ—А–Њ–≤–µ–і–µ–љ–Є–µ –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ–Њ-–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Њ–≥–Њ —Б–Ї—А–Є–љ–Є–љ–≥–∞ –љ–∞¬†–Ґ–Ґ–†-–°–Р–Я –њ–Њ–Ј–≤–Њ–ї—П–µ—В –њ–Њ–і—В–≤–µ—А–і–Є—В—М –і–Є–∞–≥–љ–Њ–Ј. –Ю–њ–Є—Б–∞–љ—Л –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ –Є¬†–њ–∞—А–∞–Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ —Е–∞—А–∞–Ї—В–µ—А–Є—Б—В–Є–Ї–Є 35¬†—Б–Є–Љ–њ—В–Њ–Љ–љ—Л—Е –љ–Њ—Б–Є—В–µ–ї–µ–є –њ–∞—В–Њ–≥–µ–љ–љ—Л—Е –Љ—Г—В–∞—Ж–Є–є –≤ –≥–µ–љ–µ —В—А–∞–љ—Б—В–Є—А–µ—В–Є–љ–∞. –Ш–љ—Б—В—А—Г–Љ–µ–љ—В–∞–ї—М–љ—Л–µ –Љ–µ—В–Њ–і—Л –Є–Љ–µ—О—В –Њ–≥—А–∞–љ–Є—З–µ–љ–Є—П –і–ї—П –њ–µ—А–≤–Є—З–љ–Њ–є –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–Є –Ґ–Ґ–†-–°–Р–Я –≤–≤–Є–і—Г –љ–µ—Б–њ–µ—Ж–Є—Д–Є—З–љ–Њ—Б—В–Є –њ–Њ–ї—Г—З–∞–µ–Љ—Л—Е —А–µ–Ј—Г–ї—М—В–∞—В–Њ–≤ –Є–ї–Є –ї–Њ–ґ–љ–Њ–Њ—В—А–Є—Ж–∞—В–µ–ї—М–љ—Л—Е –Њ—В–≤–µ—В–Њ–≤. ¬†
–Ъ–Њ–љ—Д–ї–Є–Ї—В –Є–љ—В–µ—А–µ—Б–Њ–≤. –Р–≤—В–Њ—А—Л –Ј–∞—П–≤–ї—П—О—В –Њ–±¬†–Њ—В—Б—Г—В—Б—В–≤–Є–Є –Ї–Њ–љ—Д–ї–Є–Ї—В–∞ –Є–љ—В–µ—А–µ—Б–Њ–≤.
–§–Є–љ–∞–љ—Б–Є—А–Њ–≤–∞–љ–Є–µ. –Ш—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ –њ—А–Њ–≤–Њ–і–Є–ї–Њ—Б—М –±–µ–Ј¬†—Б–њ–Њ–љ—Б–Њ—А—Б–Ї–Њ–є –њ–Њ–і–і–µ—А–ґ–Ї–Є.
–Ш–љ—Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–љ–Њ–µ —Б–Њ–≥–ї–∞—Б–Є–µ. –Я–∞—Ж–Є–µ–љ—В—Л –њ–Њ–і–њ–Є—Б–∞–ї–Є –Є–љ—Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–љ–Њ–µ —Б–Њ–≥–ї–∞—Б–Є–µ –љ–∞¬†—Г—З–∞—Б—В–Є–µ –≤¬†–Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є.
N.A. Suponeva, Corresponding member of the RASci, MD, PhD, Prof., D.A. Grishina, MD, PhD, M.S. Kazieva
FSBSI вАШResearch Center of NeurologyвАЩ
Contact person: Maria S. Kazieva, maria.kazieva555@gmail.com
Hereditary transthyretin familial amyloid polyneuropathy (TTR-FAP) is a rare, severe, steadily progressive, multisystem disease, in which the peripheral and autonomic nervous systems are most often affected. Given the availability of developed pathogenetic therapy, early detection of the pathology is extremely important.
Study objective вАУ to analyze cases of TTR-FAP identified at the Research Center of Neurology in the period 2018вАУ2025.
Material and methods. From 2018 to 2025, the Research Center of Neurology screened 750 patients with chronic polyneuropathy of unspecified genesis for TTR-FAP using a selection algorithm based on key diagnoses in combination with вАШred flagsвАЩ. In patients with a verified diagnosis of TTR-FAP, clinical, instrumental and laboratory characteristics were analyzed.
Results. Mutation in the TTR gene was detected in 35 patients (5%): 21 (60%) men and 14 (40%) women. Additional molecular genetic testing of 24 relatives of patients revealed 13 asymptomatic carriers of the mutant gene. The most common mutation was c.148G>A (Val50Met) вАУ 20 (43%) cases. The average age of patients was 52 years (minimum вАУ 23 years, maximum вАУ 81 years). The average time from the onset of the disease to diagnosis was 2.8 years. All patients (35 (100%)) were found to have polyneuritic disorders in combination with symptoms of gastrointestinal tract (22 (63%)) and cardiovascular system (12 (34%)) damage. Blood tests for NT-proBNP were taken from 11 (31%) patients. Elevated levels (over 125 pg/ml) were detected in 6 (55%) patients. Electroneuromyography of peripheral nerves revealed neurophysiological signs of generalized symmetric damage to sensory and motor nerves in 31 (89%) patients with TTR-FAP. Axonal damage to peripheral nerves was detected in 27 (87%) of 31 (89%) cases, and demyelinating damage was detected in 4 (13%) cases. Nerve ultrasound of peripheral nerves in 9 (26%) patients using the Ultrasound Pattern Sum Score protocol revealed a total score of nine or more in 3 (33%) patients, which corresponds to changes characteristic of chronic inflammatory demyelinating polyradiculoneuropathy. Changes were mild or absent in 6 (67%) patients. Of the 7 (20%) patients with TTR-FAP who underwent morphological examination of the sural nerve and subcutaneous fat, only 4 (57%) showed morphological signs of amyloidosis.
Conclusion. In the era of improving targeted pathogenetic therapy for TTR-FAP, the relevance of timely detection of the disease is extremely high. In the presence of вАШred flagsвАЩ in patients with progressive chronic polyneuropathy, molecular genetic screening for TTR-FAP allows confirming the diagnosis. Most patients with TTR-FAP have a late onset. Signs of polyneuropathy are combined with gastrointestinal, cardiovascular symptoms and unintentional weight loss. Electroneuromyography confirms the nature of the lesion of the peripheral nerves, most often primarily axonal. Ultrasound examination of the peripheral nerves and biopsy of the sural nerve in more than a third of cases do not provide information useful for diagnosing TTR-FAP.
–£–≤–∞–ґ–∞–µ–Љ—Л–є –њ–Њ—Б–µ—В–Є—В–µ–ї—М uMEDp!
–£–≤–µ–і–Њ–Љ–ї—П–µ–Љ –Т–∞—Б –Њ —В–Њ–Љ, —З—В–Њ –Ј–і–µ—Б—М —Б–Њ–і–µ—А–ґ–Є—В—Б—П –Є–љ—Д–Њ—А–Љ–∞—Ж–Є—П, –њ—А–µ–і–љ–∞–Ј–љ–∞—З–µ–љ–љ–∞—П –Є—Б–Ї–ї—О—З–Є—В–µ–ї—М–љ–Њ –і–ї—П —Б–њ–µ—Ж–Є–∞–ї–Є—Б—В–Њ–≤ –Ј–і—А–∞–≤–Њ–Њ—Е—А–∞–љ–µ–љ–Є—П.
–Х—Б–ї–Є –Т—Л –љ–µ —П–≤–ї—П–µ—В–µ—Б—М —Б–њ–µ—Ж–Є–∞–ї–Є—Б—В–Њ–Љ –Ј–і—А–∞–≤–Њ–Њ—Е—А–∞–љ–µ–љ–Є—П, –∞–і–Љ–Є–љ–Є—Б—В—А–∞—Ж–Є—П –љ–µ –љ–µ—Б–µ—В –Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ—Б—В–Є –Ј–∞ –≤–Њ–Ј–Љ–Њ–ґ–љ—Л–µ –Њ—В—А–Є—Ж–∞—В–µ–ї—М–љ—Л–µ –њ–Њ—Б–ї–µ–і—Б—В–≤–Є—П, –≤–Њ–Ј–љ–Є–Ї—И–Є–µ –≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ —Б–∞–Љ–Њ—Б—В–Њ—П—В–µ–ї—М–љ–Њ–≥–Њ –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є—П –Т–∞–Љ–Є –Є–љ—Д–Њ—А–Љ–∞—Ж–Є–Є —Б –њ–Њ—А—В–∞–ї–∞ –±–µ–Ј –њ—А–µ–і–≤–∞—А–Є—В–µ–ї—М–љ–Њ–є –Ї–Њ–љ—Б—Г–ї—М—В–∞—Ж–Є–Є —Б –≤—А–∞—З–Њ–Љ.
–Э–∞–ґ–Є–Љ–∞—П –љ–∞ –Ї–љ–Њ–њ–Ї—Г ¬Ђ–Т–Њ–є—В–Є¬ї, –Т—Л –њ–Њ–і—В–≤–µ—А–ґ–і–∞–µ—В–µ, —З—В–Њ —П–≤–ї—П–µ—В–µ—Б—М –≤—А–∞—З–Њ–Љ –Є–ї–Є —Б—В—Г–і–µ–љ—В–Њ–Љ –Љ–µ–і–Є—Ж–Є–љ—Б–Ї–Њ–≥–Њ –≤—Г–Ј–∞.