ðÆð░ÐÇð©ð░ð¢Ðéð¢Ðïð╣ Ðüð©ð¢ð┤ÐÇð¥ð╝ ð┐ðÁÐÇð▓ð©Ðçð¢ð¥ð│ð¥ ð▒ð©ð╗ð©ð░ÐÇð¢ð¥ð│ð¥ Ðàð¥ð╗ð░ð¢ð│ð©Ðéð░ Ðü┬áð┐ÐÇð©ðÀð¢ð░ð║ð░ð╝ð© ð░ÐâÐéð¥ð©ð╝ð╝Ðâð¢ð¢ð¥ð│ð¥ ð│ðÁð┐ð░Ðéð©Ðéð░: ð║ð╗ð©ð¢ð©ÐçðÁÐüð║ð¥ðÁ ð¢ð░ð▒ð╗ÐÄð┤ðÁð¢ð©ðÁ ð║ð░ð║ ð┐ð¥ð▓ð¥ð┤ ð┤ð╗ÐÅ ð┤ð©Ðüð║ÐâÐüÐüð©ð© ð¥ ðÁð┤ð©ð¢ÐüÐéð▓ðÁ ð© ð┤ð▓ð¥ð╣ÐüÐéð▓ðÁð¢ð¢ð¥ÐüÐéð© ð░ÐâÐéð¥ð©ð╝ð╝Ðâð¢ð¢ð¥ð╣ ð┐ð░Ðéð¥ð╗ð¥ð│ð©ð© ð┐ðÁÐçðÁð¢ð©
- ðÉð¢ð¢ð¥Ðéð░Ðåð©ÐÅ
- ðíÐéð░ÐéÐîÐÅ
- ðíÐüÐïð╗ð║ð©
- English

ðÆðÁð┤ðÁð¢ð©ðÁ
ðÉÐâÐéð¥ð©ð╝ð╝Ðâð¢ð¢ÐïðÁ ðÀð░ð▒ð¥ð╗ðÁð▓ð░ð¢ð©ÐÅ ð┐ðÁÐçðÁð¢ð© (ðÉðÿðùðƒ) ð┐ÐÇðÁð┤ÐüÐéð░ð▓ð╗ÐÅÐÄÐé Ðüð¥ð▒ð¥ð╣ ð│ÐÇÐâð┐ð┐Ðâ ð©ð╝ð╝Ðâð¢ð¥ð¥ð┐ð¥ÐüÐÇðÁð┤ð¥ð▓ð░ð¢ð¢ÐïÐà ð¢ð░ÐÇÐâÐêðÁð¢ð©ð╣ ÐüÐéÐÇÐâð║ÐéÐâÐÇÐï ð©┬áÐäÐâð¢ð║Ðåð©ð© ð│ðÁð┐ð░Ðéð¥Ðåð©Ðéð¥ð▓ ð©┬áðÂðÁð╗Ðçð¢ÐïÐà ð┐ÐÇð¥Ðéð¥ð║ð¥ð▓. ðÆ┬áð¥Ðüð¢ð¥ð▓ðÁ ð©Ðà ð║ð╗ð░ÐüÐüð©Ðäð©ð║ð░Ðåð©ð© ð╗ðÁðÂð©Ðé ð┐ÐÇðÁð©ð╝ÐâÐëðÁÐüÐéð▓ðÁð¢ð¢ð░ÐÅ ð╝ð©ÐêðÁð¢Ðî ð░ÐâÐéð¥ð░ð│ÐÇðÁÐüÐüð©ð©: ð▓Ðïð┤ðÁð╗ÐÅÐÄÐé ð¢ð¥ðÀð¥ð╗ð¥ð│ð©ð© Ðü┬áð┐ÐÇðÁð¥ð▒ð╗ð░ð┤ð░ð¢ð©ðÁð╝ ð│ðÁð┐ð░Ðéð¥ÐåðÁð╗ð╗ÐÄð╗ÐÅÐÇð¢ð¥ð│ð¥ ð┐ð¥ÐÇð░ðÂðÁð¢ð©ÐÅ (ð░ÐâÐéð¥ð©ð╝ð╝Ðâð¢ð¢Ðïð╣ ð│ðÁð┐ð░Ðéð©Ðé) ð©┬áÐü ð┐ÐÇðÁð¥ð▒ð╗ð░ð┤ð░ð¢ð©ðÁð╝ Ðàð¥ð╗ð░ð¢ð│ð©ð¥ÐåðÁð╗ð╗ÐÄð╗ÐÅÐÇð¢ð¥ð│ð¥ ð┐ð¥ÐÇð░ðÂðÁð¢ð©ÐÅ (ð┐ðÁÐÇð▓ð©Ðçð¢Ðïð╣ ð▒ð©ð╗ð©ð░ÐÇð¢Ðïð╣ Ðàð¥ð╗ð░ð¢ð│ð©Ðé, ðƒðæðÑ, ð©┬áð┐ðÁÐÇð▓ð©Ðçð¢Ðïð╣ Ðüð║ð╗ðÁÐÇð¥ðÀð©ÐÇÐâÐÄÐëð©ð╣ Ðàð¥ð╗ð░ð¢ð│ð©Ðé, ðƒðíðÑ). ðÆ┬áð¥Ðéð┤ðÁð╗Ðîð¢ÐâÐÄ ð║ð░ÐéðÁð│ð¥ÐÇð©ÐÄ ð╝ð¥ðÂð¢ð¥ ð▓Ðïð¢ðÁÐüÐéð© ð▓ð░ÐÇð©ð░ð¢Ðéð¢ÐïðÁ Ðüð©ð¢ð┤ÐÇð¥ð╝Ðï, ð┐ÐÇð© ð║ð¥Ðéð¥ÐÇÐïÐà Ðâ┬áð┐ð░Ðåð©ðÁð¢Ðéð░ ð¥ð┤ð¢ð¥ð▓ÐÇðÁð╝ðÁð¢ð¢ð¥ ð©ð╗ð© ð┐ð¥Ðüð╗ðÁð┤ð¥ð▓ð░ÐéðÁð╗Ðîð¢ð¥ ð▓ÐïÐÅð▓ð╗ðÁð¢Ðï ÐçðÁÐÇÐéÐï ð┤ð▓ÐâÐà ð©┬áð▒ð¥ð╗ðÁðÁ ðÉðÿðùðƒ [1].
ðÉÐâÐéð¥ð©ð╝ð╝Ðâð¢ð¢Ðïð╣ ð│ðÁð┐ð░Ðéð©Ðé (ðÉðÿðô) ÔÇô ÐìÐéð¥ ÐàÐÇð¥ð¢ð©ÐçðÁÐüð║ð¥ðÁ ð┐ÐÇð¥ð│ÐÇðÁÐüÐüð©ÐÇÐâÐÄÐëðÁðÁ ð▓ð¥Ðüð┐ð░ð╗ð©ÐéðÁð╗Ðîð¢ð¥ðÁ ðÀð░ð▒ð¥ð╗ðÁð▓ð░ð¢ð©ðÁ ð┐ðÁÐçðÁð¢ð© ð¢ðÁð©ðÀð▓ðÁÐüÐéð¢ð¥ð╣ ÐìÐéð©ð¥ð╗ð¥ð│ð©ð©, Ðàð░ÐÇð░ð║ÐéðÁÐÇð©ðÀÐâÐÄÐëðÁðÁÐüÐÅ ð│ðÁÐéðÁÐÇð¥ð│ðÁð¢ð¢ð¥ÐüÐéÐîÐÄ ð║ð╗ð©ð¢ð©ð║ð¥-ð╗ð░ð▒ð¥ÐÇð░Ðéð¥ÐÇð¢ÐïÐà ð©┬áð╝ð¥ÐÇÐäð¥ð╗ð¥ð│ð©ÐçðÁÐüð║ð©Ðà ð┐ÐÇð¥ÐÅð▓ð╗ðÁð¢ð©ð╣, ð▓┬áð¥Ðüð¢ð¥ð▓ðÁ ð║ð¥Ðéð¥ÐÇð¥ð│ð¥ ð╗ðÁðÂð░Ðé ð╝ðÁÐàð░ð¢ð©ðÀð╝Ðï ð░ÐâÐéð¥ð░ð│ÐÇðÁÐüÐüð©ð©; ð▒ðÁðÀ ð╗ðÁÐçðÁð¢ð©ÐÅ ð┐ÐÇð©ð▓ð¥ð┤ð©Ðé ð║┬áÐäð¥ÐÇð╝ð©ÐÇð¥ð▓ð░ð¢ð©ÐÄ ð▓ÐïÐÇð░ðÂðÁð¢ð¢ð¥ð│ð¥ Ðäð©ð▒ÐÇð¥ðÀð░ ð©┬áÐåð©ÐÇÐÇð¥ðÀð░ ð┐ðÁÐçðÁð¢ð© [2].
ðƒðÁÐÇð▓ð©Ðçð¢Ðïð╣ ð▒ð©ð╗ð©ð░ÐÇð¢Ðïð╣ Ðàð¥ð╗ð░ð¢ð│ð©Ðé ÔÇô ÐàÐÇð¥ð¢ð©ÐçðÁÐüð║ð¥ðÁ ð©ð╝ð╝Ðâð¢ð¥ð¥ð┐ð¥ÐüÐÇðÁð┤ð¥ð▓ð░ð¢ð¢ð¥ðÁ ðÀð░ð▒ð¥ð╗ðÁð▓ð░ð¢ð©ðÁ ð┐ðÁÐçðÁð¢ð©, ð┐ÐÇð¥ÐéðÁð║ð░ÐÄÐëðÁðÁ Ðü┬áð┤ðÁÐüÐéÐÇÐâð║Ðåð©ðÁð╣ ð╝ðÁð╗ð║ð©Ðà ðÂðÁð╗Ðçð¢ÐïÐà ð┐ÐÇð¥Ðéð¥ð║ð¥ð▓, Ðàð░ÐÇð░ð║ÐéðÁÐÇð¢ð¥ð╣ ÐçðÁÐÇÐéð¥ð╣ ÐÅð▓ð╗ÐÅðÁÐéÐüÐÅ Ðüð©ð¢ð┤ÐÇð¥ð╝ Ðàð¥ð╗ðÁÐüÐéð░ðÀð░ [3ÔÇô5].
ðíð¥ÐçðÁÐéð░ð¢ð©ðÁ ð┐ÐÇð©ðÀð¢ð░ð║ð¥ð▓ ðÉðÿðô ð©┬áðƒðæðÑ Ðâ┬áð¥ð┤ð¢ð¥ð│ð¥ ð┐ð░Ðåð©ðÁð¢Ðéð░ ð¥Ðéð¢ð¥ÐüÐÅÐé ð║┬áð¥Ðüð¥ð▒Ðïð╝ Ðäð¥ÐÇð╝ð░ð╝ ðÉðÿðùðƒ; ð©Ðà ð▓ð║ð╗ð░ð┤ ð▓┬áð║ð╗ð©ð¢ð©ÐçðÁÐüð║ÐâÐÄ ð║ð░ÐÇÐéð©ð¢Ðâ ð╝ð¥ðÂðÁÐé ð▒ÐïÐéÐî ÐÇð░ð▓ð¢ð¥ðÀð¢ð░Ðçð¢Ðïð╝, ð╗ð©ð▒ð¥ ð╝ð¥ðÂðÁÐé ð┤ð¥ð╝ð©ð¢ð©ÐÇð¥ð▓ð░ÐéÐî ð¥ð┤ð©ð¢ ð©ðÀ┬áð║ð¥ð╝ð┐ð¥ð¢ðÁð¢Ðéð¥ð▓, ÐçÐéð¥ Ðüð¥ðÀð┤ð░ðÁÐé ðÀð¢ð░Ðçð©ÐéðÁð╗Ðîð¢ÐâÐÄ ð▓ð░ÐÇð©ð░ð▒ðÁð╗Ðîð¢ð¥ÐüÐéÐî ð▓┬áÐéðÁÐçðÁð¢ð©ð© ð▒ð¥ð╗ðÁðÀð¢ð©. ðÆð¥ð┐ÐÇð¥Ðü ð¥┬áð┐ð¥Ðüð╗ðÁð┤ð¥ð▓ð░ÐéðÁð╗Ðîð¢ð¥ÐüÐéð© ð╝ð░ð¢ð©ÐäðÁÐüÐéð░Ðåð©ð© ð║ð¥ð╝ð┐ð¥ð¢ðÁð¢Ðéð¥ð▓ ð¥Ðüð¥ð▒ÐïÐà Ðäð¥ÐÇð╝ ðÉðÿðùðƒ ð¥ÐüÐéð░ðÁÐéÐüÐÅ ð┤ð©Ðüð║ÐâÐüÐüð©ð¥ð¢ð¢Ðïð╝ [3], ÐéðÁÐÇð╝ð©ð¢ð¥ð╗ð¥ð│ð©ÐÅ ð©┬áð¢ð¥ð╝ðÁð¢ð║ð╗ð░ÐéÐâÐÇð░ ð¥ð║ð¥ð¢Ðçð░ÐéðÁð╗Ðîð¢ð¥ ð¢ðÁ┬áÐüÐéð░ð¢ð┤ð░ÐÇÐéð©ðÀð©ÐÇð¥ð▓ð░ð¢Ðï: ð▓┬áÐÇÐâÐüÐüð║ð¥ÐÅðÀÐïÐçð¢ð¥ð╣ ð╝ðÁð┤ð©Ðåð©ð¢Ðüð║ð¥ð╣ ð╗ð©ÐéðÁÐÇð░ÐéÐâÐÇðÁ Ðçð░ÐüÐéð¥ ÐéÐÇð░ð┤ð©Ðåð©ð¥ð¢ð¢ð¥ ð©Ðüð┐ð¥ð╗ÐîðÀÐâðÁÐéÐüÐÅ ð┐ð¥ð¢ÐÅÐéð©ðÁ ┬½ð░ÐâÐéð¥ð©ð╝ð╝Ðâð¢ð¢Ðïð╣ ð┐ðÁÐÇðÁð║ÐÇðÁÐüÐé┬╗, ð▓┬áÐìÐéð¥ð╝ Ðüð╗ÐâÐçð░ðÁ ð▓┬áð┤ð©ð░ð│ð¢ð¥ðÀðÁ ð┐ðÁÐÇðÁÐçð©Ðüð╗ÐÅÐÄÐéÐüÐÅ ð┤ð▓ð░ (ð©ð╗ð© ÐéÐÇð©) ð©ð╝ðÁÐÄÐëð©ÐàÐüÐÅ Ðâ┬áð┐ð░Ðåð©ðÁð¢Ðéð░ ðÉðÿðùðƒ, ð¢ð░ð┐ÐÇð©ð╝ðÁÐÇ, ┬½Ðüð©ð¢ð┤ÐÇð¥ð╝ ð░ÐâÐéð¥ð©ð╝ð╝Ðâð¢ð¢ð¥ð│ð¥ ð┐ðÁÐÇðÁð║ÐÇðÁÐüÐéð░: ðÉðÿðô/ðƒðæðÑ┬╗. ð×ð┤ð¢ð░ð║ð¥ ÐìÐéð░ Ðäð¥ÐÇð╝Ðâð╗ð©ÐÇð¥ð▓ð║ð░ ð¢ðÁ┬áð┤ð░ðÁÐé ð┐ÐÇðÁð┤ÐüÐéð░ð▓ð╗ðÁð¢ð©ÐÅ ð¥┬áÐéð¥ð╝, ð║ð░ð║ð¥ðÁ ðÀð░ð▒ð¥ð╗ðÁð▓ð░ð¢ð©ðÁ ð┐ÐÇðÁð¥ð▒ð╗ð░ð┤ð░ðÁÐé ð▓┬áð║ð¥ð¢ð║ÐÇðÁÐéð¢ð¥ð╝ Ðüð╗ÐâÐçð░ðÁ; ð┐ð¥ÐìÐéð¥ð╝Ðâ ðÁÐëðÁ ð▓┬á2015 ð│.┬áðòð▓ÐÇð¥ð┐ðÁð╣Ðüð║ð¥ð╣ ð░ÐüÐüð¥Ðåð©ð░Ðåð©ðÁð╣ ð┐ð¥┬áð©ðÀÐâÐçðÁð¢ð©ÐÄ ð▒ð¥ð╗ðÁðÀð¢ðÁð╣ ð┐ðÁÐçðÁð¢ð© (EASL) ð▒Ðïð╗ð¥ ÐÇðÁð║ð¥ð╝ðÁð¢ð┤ð¥ð▓ð░ð¢ð¥ ð¥ð▒ð¥ðÀð¢ð░Ðçð░ÐéÐî ð▓ð░ÐÇð©ð░ð¢Ðéð¢ÐïðÁ Ðäð¥ÐÇð╝Ðï ð┐ð¥┬áð¥Ðüð¢ð¥ð▓ð¢ð¥ð╝Ðâ ðÀð░ð▒ð¥ð╗ðÁð▓ð░ð¢ð©ÐÄ, ð┤ð¥ð╝ð©ð¢ð©ÐÇÐâÐÄÐëðÁð╝Ðâ ð▓┬áð║ð╗ð©ð¢ð©ÐçðÁÐüð║ð¥ð╣ ð║ð░ÐÇÐéð©ð¢ðÁ, Ðü┬áð┤ð¥ð┐ð¥ð╗ð¢ðÁð¢ð©ðÁð╝, Ðâð║ð░ðÀÐïð▓ð░ÐÄÐëð©ð╝ ð¢ð░┬áð┐ÐÇð©ðÀð¢ð░ð║ð© ð▓Ðéð¥ÐÇð¥ð│ð¥ ðÉðÿðùðƒ. ð¡Ðéð¥ ð▒ð¥ð╗ðÁðÁ Ðéð¥Ðçð¢ð¥ ð¥ÐéÐÇð░ðÂð░ðÁÐé ÐüÐâÐéÐî ð┐ÐÇð¥ð©ÐüÐàð¥ð┤ÐÅÐëðÁð│ð¥ ð┐ð░Ðéð¥ð╗ð¥ð│ð©ÐçðÁÐüð║ð¥ð│ð¥ ð┐ÐÇð¥ÐåðÁÐüÐüð░, ð¢ð░ð┐ÐÇð©ð╝ðÁÐÇ, ð▓┬áÐüð╗ÐâÐçð░ðÁ Ðüð¥ÐçðÁÐéð░ð¢ð©ÐÅ ð▓ÐïÐÇð░ðÂðÁð¢ð¢ð¥ð│ð¥ Ðàð¥ð╗ðÁÐüÐéð░Ðéð©ÐçðÁÐüð║ð¥ð│ð¥ Ðüð©ð¢ð┤ÐÇð¥ð╝ð░ Ðü┬áð¢ð©ðÀð║ð¥ð╣ ð©ð╗ð© Ðâð╝ðÁÐÇðÁð¢ð¢ð¥ð╣ ð░ð║Ðéð©ð▓ð¢ð¥ÐüÐéÐîÐÄ ð│ðÁð┐ð░Ðéð©Ðéð░ ÐÇðÁð║ð¥ð╝ðÁð¢ð┤ð¥ð▓ð░ð¢ð¥ Ðäð¥ÐÇð╝Ðâð╗ð©ÐÇð¥ð▓ð░ÐéÐî ð┤ð©ð░ð│ð¢ð¥ðÀ ð║ð░ð║ ┬½ðƒðæðÑ Ðü┬áð┐ÐÇð©ðÀð¢ð░ð║ð░ð╝ð© ðÉðÿðô┬╗ [6].
ðÆÐüðÁð╝ ð┐ð░Ðåð©ðÁð¢Ðéð░ð╝ Ðü┬áð┐ð¥ð┤ð¥ðÀÐÇðÁð¢ð©ðÁð╝ ð¢ð░┬áð▓ð░ÐÇð©ð░ð¢Ðéð¢ÐâÐÄ Ðäð¥ÐÇð╝Ðâ ðƒðæðÑ ÐÇðÁð║ð¥ð╝ðÁð¢ð┤ÐâðÁÐéÐüÐÅ ð▓Ðïð┐ð¥ð╗ð¢ðÁð¢ð©ðÁ ð┐Ðâð¢ð║Ðåð©ð¥ð¢ð¢ð¥ð╣ ð▒ð©ð¥ð┐Ðüð©ð© ð┐ðÁÐçðÁð¢ð© (ð┐ÐÇð© ð¥ÐéÐüÐâÐéÐüÐéð▓ð©ð© ð┐ÐÇð¥Ðéð©ð▓ð¥ð┐ð¥ð║ð░ðÀð░ð¢ð©ð╣) [3ÔÇô5]. ðÆ┬áð┤ð░ð╗Ðîð¢ðÁð╣ÐêðÁð╝ ð┤ð©ð░ð│ð¢ð¥ÐüÐéð©ð║ð░ ð▓ð░ÐÇð©ð░ð¢Ðéð¢ð¥ð│ð¥ Ðüð©ð¢ð┤ÐÇð¥ð╝ð░ ð┐ÐÇð¥ð▓ð¥ð┤ð©ÐéÐüÐÅ Ðü┬áð┐ÐÇð©ð╝ðÁð¢ðÁð¢ð©ðÁð╝ Ðéð░ð║ ð¢ð░ðÀÐïð▓ð░ðÁð╝ÐïÐà ðƒð░ÐÇð©ðÂÐüð║ð©Ðà ð║ÐÇð©ÐéðÁÐÇð©ðÁð▓, ð║ð¥Ðéð¥ÐÇÐïðÁ ð¥ð┤ð¥ð▒ÐÇðÁð¢Ðï ðòð▓ÐÇð¥ð┐ðÁð╣Ðüð║ð¥ð╣ ð░ÐüÐüð¥Ðåð©ð░Ðåð©ðÁð╣ ð┐ð¥┬áð©ðÀÐâÐçðÁð¢ð©ÐÄ ð▒ð¥ð╗ðÁðÀð¢ðÁð╣ ð┐ðÁÐçðÁð¢ð© ð©┬áÐÇð¥ÐüÐüð©ð╣Ðüð║ð©ð╝ð© Ðìð║Ðüð┐ðÁÐÇÐéð░ð╝ð© [3]; ÐéðÁð╝ ð¢ðÁ┬áð╝ðÁð¢ðÁðÁ ð▓ð▓ð©ð┤Ðâ Ðüð╗ð¥ðÂð¢ð¥ÐüÐéð© ð┐ð░Ðéð¥ð│ðÁð¢ðÁðÀð░ ð©┬áð¥ÐéÐüÐâÐéÐüÐéð▓ð©ÐÅ ÐüÐéð░ð¢ð┤ð░ÐÇÐéð©ðÀð©ÐÇð¥ð▓ð░ð¢ð¢ð¥ð│ð¥ ð┐ð¥ð┤Ðàð¥ð┤ð░ ÐçÐâð▓ÐüÐéð▓ð©ÐéðÁð╗Ðîð¢ð¥ÐüÐéÐî ð©┬áÐüð┐ðÁÐåð©Ðäð©Ðçð¢ð¥ÐüÐéÐî ð║ÐÇð©ÐéðÁÐÇð©ðÁð▓ ÐéÐÇðÁð▒ÐâðÁÐé ð┐ðÁÐÇðÁÐüð╝ð¥ÐéÐÇð░ [7]. ðƒÐÇð© ð▓ÐïÐÅð▓ð╗ðÁð¢ð©ð© ð┐ÐÇð©ðÀð¢ð░ð║ð¥ð▓ ðÉðÿðô ð▓┬áð┤ð¥ð┐ð¥ð╗ð¢ðÁð¢ð©ðÁ ð║┬áÐüÐéð░ð¢ð┤ð░ÐÇÐéð¢ð¥ð╝Ðâ ð╗ðÁÐçðÁð¢ð©ÐÄ ðƒðæðÑ ð┐ÐÇðÁð┐ð░ÐÇð░Ðéð░ð╝ð© ÐâÐÇÐüð¥ð┤ðÁðÀð¥ð║Ðüð©Ðàð¥ð╗ðÁð▓ð¥ð╣ ð║ð©Ðüð╗ð¥ÐéÐï (ðúðöðÑðÜ) ÐéÐÇðÁð▒ÐâðÁÐéÐüÐÅ ð¢ð░ðÀð¢ð░ÐçðÁð¢ð©ðÁ ð©ð╝ð╝Ðâð¢ð¥ÐüÐâð┐ÐÇðÁÐüÐüð©ð▓ð¢ð¥ð╣ ÐéðÁÐÇð░ð┐ð©ð© (ðÿðíðó) [3, 6].
ðÜð╗ð©ð¢ð©ÐçðÁÐüð║ð©ð╣ Ðüð╗ÐâÐçð░ð╣
ðÆ┬áð║ð░ÐçðÁÐüÐéð▓ðÁ ð©ð╗ð╗ÐÄÐüÐéÐÇð░Ðåð©ð© Ðüð╗ð¥ðÂð¢ð¥ÐüÐéð© ð▓ðÁð┤ðÁð¢ð©ÐÅ ð┐ð░Ðåð©ðÁð¢Ðéð¥ð▓ Ðü┬áð░ÐâÐéð¥ð©ð╝ð╝Ðâð¢ð¢Ðïð╝ ð▓ð░ÐÇð©ð░ð¢Ðéð¢Ðïð╝ Ðüð©ð¢ð┤ÐÇð¥ð╝ð¥ð╝ ð╝ð¥ðÂðÁÐé Ðüð╗ÐâðÂð©ÐéÐî ð║ð╗ð©ð¢ð©ÐçðÁÐüð║ð¥ðÁ ð¢ð░ð▒ð╗ÐÄð┤ðÁð¢ð©ðÁ ð┐ð░Ðåð©ðÁð¢Ðéð║ð© ðø. Ðü┬áð┤ðÁð▒ÐÄÐéð¥ð╝ ðÀð░ð▒ð¥ð╗ðÁð▓ð░ð¢ð©ÐÅ ð▓┬áð▓ð¥ðÀÐÇð░ÐüÐéðÁ 45 ð╗ðÁÐé, ð║ð¥Ðéð¥ÐÇð░ÐÅ ð┐ÐÇðÁð┤ÐèÐÅð▓ð╗ÐÅð╗ð░ ðÂð░ð╗ð¥ð▒Ðï ð¢ð░┬áðÀÐâð┤ ð║ð¥ðÂð©, ð│ð¥ÐÇðÁÐçÐî ð▓ð¥ ÐÇÐéÐâ, ð┐ðÁÐÇð©ð¥ð┤ð©ÐçðÁÐüð║ÐâÐÄ Ðéð¥Ðêð¢ð¥ÐéÐâ.
ðƒð░Ðåð©ðÁð¢Ðéð║ð░ ð▒Ðïð╗ð░ ð┐ÐÇð¥ð©ð¢Ðäð¥ÐÇð╝ð©ÐÇð¥ð▓ð░ð¢ð░ ð¥┬áÐåðÁð╗ÐÅÐà ð©ÐüÐüð╗ðÁð┤ð¥ð▓ð░ð¢ð©ÐÅ ð©┬áð┤ð░ð╗ð░ Ðüð¥ð│ð╗ð░Ðüð©ðÁ ð¢ð░┬áð┐Ðâð▒ð╗ð©ð║ð░Ðåð©ÐÄ ð┤ð░ð¢ð¢ÐïÐà ð©ÐüÐéð¥ÐÇð©ð© ð▒ð¥ð╗ðÁðÀð¢ð© ð▓┬áð¥ð▒ðÁðÀð╗ð©ÐçðÁð¢ð¢ð¥ð╝ ð▓ð©ð┤ðÁ; ð¥Ðüð╝ð¥ÐéÐÇ, ð¥ð▒Ðüð╗ðÁð┤ð¥ð▓ð░ð¢ð©ðÁ ð©┬áð╗ðÁÐçðÁð¢ð©ðÁ ð┐ÐÇð¥ð▓ðÁð┤ðÁð¢Ðï Ðüð¥ð│ð╗ð░Ðüð¢ð¥ ð║ð╗ð©ð¢ð©ÐçðÁÐüð║ð©ð╝ ÐÇðÁð║ð¥ð╝ðÁð¢ð┤ð░Ðåð©ÐÅð╝ ð£ð©ð¢ð©ÐüÐéðÁÐÇÐüÐéð▓ð░ ðÀð┤ÐÇð░ð▓ð¥ð¥ÐàÐÇð░ð¢ðÁð¢ð©ÐÅ ðáðñ.
ðÉð¢ð░ð╝ð¢ðÁðÀ ðÂð©ðÀð¢ð©
ðƒð░Ðåð©ðÁð¢Ðé ÔÇô ðÂðÁð¢Ðëð©ð¢ð░ 47 ð╗ðÁÐé, ð┐ÐÇðÁð┐ð¥ð┤ð░ð▓ð░ÐéðÁð╗Ðî, ð┐ÐÇð¥ÐäðÁÐüÐüð©ð¥ð¢ð░ð╗Ðîð¢ÐïðÁ ð▓ÐÇðÁð┤ð¢ÐïðÁ Ðäð░ð║Ðéð¥ÐÇÐï ð¥ÐéÐüÐâÐéÐüÐéð▓ÐâÐÄÐé, ð▓ÐÇðÁð┤ð¢ÐïðÁ ð┐ÐÇð©ð▓ÐïÐçð║ð© ð¥ÐéÐÇð©Ðåð░ðÁÐé. ðíðÁð╝ðÁð╣ð¢Ðïð╣ ð░ð¢ð░ð╝ð¢ðÁðÀ ð¢ðÁ┬áð¥ÐéÐÅð│ð¥ÐëðÁð¢, ð╗ðÁð║ð░ÐÇÐüÐéð▓ðÁð¢ð¢ÐïðÁ ð┐ÐÇðÁð┐ð░ÐÇð░ÐéÐï ð¢ðÁ┬áð┐ÐÇð©ð¢ð©ð╝ð░ðÁÐé.
ðÉð¢ð░ð╝ð¢ðÁðÀ ðÀð░ð▒ð¥ð╗ðÁð▓ð░ð¢ð©ÐÅ
ðíÐçð©Ðéð░ðÁÐé ÐüðÁð▒ÐÅ ð▒ð¥ð╗Ðîð¢ð¥ð╣ Ðü┬á45-ð╗ðÁÐéð¢ðÁð│ð¥ ð▓ð¥ðÀÐÇð░ÐüÐéð░, ð║ð¥ð│ð┤ð░ ð¢ð░┬áÐäð¥ð¢ðÁ ð┐ð¥ð╗ð¢ð¥ð│ð¥ ðÀð┤ð¥ÐÇð¥ð▓ÐîÐÅ ð▓ð┐ðÁÐÇð▓ÐïðÁ ð¥Ðéð╝ðÁÐéð©ð╗ð░ ð┐ð¥ÐÅð▓ð╗ðÁð¢ð©ðÁ ð┐ðÁÐÇð©ð¥ð┤ð©ÐçðÁÐüð║ð¥ð│ð¥ ð║ð¥ðÂð¢ð¥ð│ð¥ ðÀÐâð┤ð░ ð▓┬áð¥ð▒ð╗ð░ÐüÐéð© ð╗ð░ð┤ð¥ð¢ðÁð╣, ð│ð¥ð╗ðÁð¢ðÁð╣ ð©┬áÐüÐéð¥ð┐: ð┐ÐÇðÁð┤ð┐ÐÇð©ð¢ð©ð╝ð░ð╗ð©ÐüÐî ð┐ð¥ð┐ÐïÐéð║ð© ÐéðÁÐÇð░ð┐ð©ð© ð▒ð╗ð¥ð║ð░Ðéð¥ÐÇð░ð╝ð© ð│ð©ÐüÐéð░ð╝ð©ð¢ð¥ð▓ÐïÐà ðØ1-ÐÇðÁÐåðÁð┐Ðéð¥ÐÇð¥ð▓ (ð╗ð¥ÐÇð░Ðéð░ð┤ð©ð¢) ð©┬áÐìð╝ð¥ð╗ðÁð¢Ðéð░ð╝ð© Ðü┬áð¢ðÁðÀð¢ð░Ðçð©ÐéðÁð╗Ðîð¢Ðïð╝ ÐìÐäÐäðÁð║Ðéð¥ð╝. ðíð┐ÐâÐüÐéÐÅ ð│ð¥ð┤ ÐüÐéð░ð╗ð░ ð¥Ðéð╝ðÁÐçð░ÐéÐî ð│ð¥ÐÇðÁÐçÐî ð▓ð¥ ÐÇÐéÐâ ð┐ð¥┬áÐâÐéÐÇð░ð╝, ð┐ðÁÐÇð©ð¥ð┤ð©ÐçðÁÐüð║ð© Ðéð¥Ðêð¢ð¥ÐéÐâ ð▒ðÁðÀ ÐçðÁÐéð║ð¥ð╣ Ðüð▓ÐÅðÀð© Ðü┬áð┐ÐÇð©ðÁð╝ð¥ð╝ ð┐ð©Ðëð©.
ðÆ┬á2019 ð│.┬áð┐ð░Ðåð©ðÁð¢Ðéð║ð░ ð▓ð┐ðÁÐÇð▓ÐïðÁ ð░ð╝ð▒Ðâð╗ð░Ðéð¥ÐÇð¢ð¥ ð¥ð▒ÐÇð░Ðéð©ð╗ð░ÐüÐî ðÀð░ ð╝ðÁð┤ð©Ðåð©ð¢Ðüð║ð¥ð╣ ð┐ð¥ð╝ð¥ÐëÐîÐÄ ð║┬áð│ð░ÐüÐéÐÇð¥Ðìð¢ÐéðÁÐÇð¥ð╗ð¥ð│Ðâ ð┤ð╗ÐÅ ð¥ð▒Ðüð╗ðÁð┤ð¥ð▓ð░ð¢ð©ÐÅ, ð┐ð¥┬áÐÇðÁðÀÐâð╗ÐîÐéð░Ðéð░ð╝ ð║ð¥Ðéð¥ÐÇð¥ð│ð¥ ð┐ð¥ð╗ÐâÐçðÁð¢Ðï Ðüð╗ðÁð┤ÐâÐÄÐëð©ðÁ ÐÇðÁðÀÐâð╗ÐîÐéð░ÐéÐï: ð▓┬áð║ð╗ð©ð¢ð©ÐçðÁÐüð║ð¥ð╝ ð░ð¢ð░ð╗ð©ðÀðÁ ð║ÐÇð¥ð▓ð© ð¥Ðéð║ð╗ð¥ð¢ðÁð¢ð©ð╣ ð¢ðÁ┬áð¥Ðéð╝ðÁÐçð░ð╗ð¥ÐüÐî; ð▓┬áð▒ð©ð¥Ðàð©ð╝ð©ÐçðÁÐüð║ð¥ð╝ ð░ð¢ð░ð╗ð©ðÀðÁ ð║ÐÇð¥ð▓ð© ð░ð╗ð░ð¢ð©ð¢ð¥ð▓ð░ÐÅ ð░ð╝ð©ð¢ð¥ÐéÐÇð░ð¢ÐüÐäðÁÐÇð░ðÀð░ (ðÉðøðó) ð┐ÐÇðÁð▓ÐïÐêð░ð╗ð░ ð▓ðÁÐÇÐàð¢ÐÄÐÄ ð│ÐÇð░ð¢ð©ÐåÐâ ð¢ð¥ÐÇð╝Ðï (ðÆðôðØ) ð▓┬á5 ÐÇð░ðÀ, ð░Ðüð┐ð░ÐÇð░ð│ð©ð¢ð¥ð▓ð░ÐÅ ð░ð╝ð©ð¢ð¥ÐéÐÇð░ð¢ÐüÐäðÁÐÇð░ðÀð░ (ðÉðíðó) ÔÇô ð▓┬á4,5 ÐÇð░ðÀð░; ð¥Ðéð╝ðÁÐçð░ð╗ÐüÐÅ ð╗ð░ð▒ð¥ÐÇð░Ðéð¥ÐÇð¢Ðïð╣ Ðüð©ð¢ð┤ÐÇð¥ð╝ Ðàð¥ð╗ðÁÐüÐéð░ðÀð░ Ðü┬áð┐ð¥ð▓ÐïÐêðÁð¢ð©ðÁð╝ ð│ð░ð╝ð╝ð░-ð│ð╗ÐâÐéð░ð╝ð©ð╗ÐéÐÇð░ð¢Ðüð┐ðÁð┐Ðéð©ð┤ð░ðÀÐï (ðôðôðóðƒ) ð┤ð¥ 7 ðÆðôðØ, ÐëðÁð╗ð¥Ðçð¢ð¥ð╣ Ðäð¥ÐüÐäð░Ðéð░ðÀÐï (ð®ðñ) ð┤ð¥ 3 ðÆðôðØ, ð│ð©ð┐ðÁÐÇÐàð¥ð╗ðÁÐüÐéðÁÐÇð©ð¢ðÁð╝ð©ÐÅ. ð×Ðüð¢ð¥ð▓ð¢ÐïðÁ ÐÇðÁðÀÐâð╗ÐîÐéð░ÐéÐï ð╗ð░ð▒ð¥ÐÇð░Ðéð¥ÐÇð¢ÐïÐà ð©ÐüÐüð╗ðÁð┤ð¥ð▓ð░ð¢ð©ð╣ ð┐ÐÇðÁð┤ÐüÐéð░ð▓ð╗ðÁð¢Ðï ð▓┬áÐéð░ð▒ð╗. 1.
ðƒð░Ðåð©ðÁð¢Ðéð║ð░ ð¢ð░ð▒ð╗ÐÄð┤ð░ð╗ð░ÐüÐî Ðâ┬áð│ð░ÐüÐéÐÇð¥Ðìð¢ÐéðÁÐÇð¥ð╗ð¥ð│ð░ ð░ð╝ð▒Ðâð╗ð░Ðéð¥ÐÇð¢ð¥: Ðüð¥ÐüÐéð¥ÐÅð¢ð©ðÁ ÐÇð░ÐüÐåðÁð¢ð©ð▓ð░ð╗ð¥ÐüÐî ð║ð░ð║ ÐàÐÇð¥ð¢ð©ÐçðÁÐüð║ð©ð╣ ð│ðÁð┐ð░Ðéð©Ðé ð¢ðÁÐâÐéð¥Ðçð¢ðÁð¢ð¢ð¥ð╣ ÐìÐéð©ð¥ð╗ð¥ð│ð©ð©; ð¢ðÁð¥ð┤ð¢ð¥ð║ÐÇð░Ðéð¢ð¥ ð┐ÐÇð¥ð▓ð¥ð┤ð©ð╗ð©ÐüÐî ð║ÐâÐÇÐüÐï ð│ðÁð┐ð░Ðéð¥ð┐ÐÇð¥ÐéðÁð║Ðéð©ð▓ð¢ð¥ð╣ ÐéðÁÐÇð░ð┐ð©ð© ð┐ÐÇðÁð┐ð░ÐÇð░Ðéð░ð╝ð© ð░ð┤ðÁð╝ðÁÐéð©ð¥ð¢ð©ð¢ð░ ð©┬áÐìÐüÐüðÁð¢Ðåð©ð░ð╗Ðîð¢ÐïÐà Ðäð¥ÐüÐäð¥ð╗ð©ð┐ð©ð┤ð¥ð▓ Ðü┬áð¢ðÁðÀð¢ð░Ðçð©ÐéðÁð╗Ðîð¢Ðïð╝ ð║ð╗ð©ð¢ð©ÐçðÁÐüð║ð©ð╝ ÐìÐäÐäðÁð║Ðéð¥ð╝ ð▓┬áð▓ð©ð┤ðÁ Ðâð╝ðÁð¢ÐîÐêðÁð¢ð©ÐÅ Ðéð¥Ðêð¢ð¥ÐéÐï ð©┬áð┐ðÁÐÇð©ð¥ð┤ð©ÐçðÁÐüð║ð¥ð│ð¥ Ðüð¢ð©ðÂðÁð¢ð©ÐÅ ð©ð¢ÐéðÁð¢Ðüð©ð▓ð¢ð¥ÐüÐéð© ð║ð¥ðÂð¢ð¥ð│ð¥ ðÀÐâð┤ð░; ð╗ð░ð▒ð¥ÐÇð░Ðéð¥ÐÇð¢ÐïðÁ ð©ðÀð╝ðÁð¢ðÁð¢ð©ÐÅ Ðüð¥ÐàÐÇð░ð¢ÐÅð╗ð©ÐüÐî (Ðéð░ð▒ð╗.┬á1).
ðÆ┬áð¢ð¥ÐÅð▒ÐÇðÁ 2020 ð│.┬áð┐ð░Ðåð©ðÁð¢Ðéð║ðÁ ð▓ð┐ðÁÐÇð▓ÐïðÁ ð▓Ðïð┐ð¥ð╗ð¢ðÁð¢ ð░ð¢ð░ð╗ð©ðÀ ð║ÐÇð¥ð▓ð© ð¢ð░┬áð░ð¢Ðéð©ð╝ð©Ðéð¥Ðàð¥ð¢ð┤ÐÇð©ð░ð╗Ðîð¢ÐïðÁ ð░ÐâÐéð¥ð░ð¢Ðéð©ÐéðÁð╗ð░ (ðÉð£ðÉ-ð£2), ð║ð¥Ðéð¥ÐÇÐïðÁ ð▒Ðïð╗ð© ð▓ÐïÐÅð▓ð╗ðÁð¢Ðï ð▓┬áð┐ð¥ð▓ÐïÐêðÁð¢ð¢ð¥ð╝ Ðéð©ÐéÐÇðÁ, ð¢ð░┬áð¥Ðüð¢ð¥ð▓ð░ð¢ð©ð© ÐçðÁð│ð¥ (ð▓┬áÐüð¥ÐçðÁÐéð░ð¢ð©ð© Ðü┬áÐüð©ð¢ð┤ÐÇð¥ð╝ð¥ð╝ Ðàð¥ð╗ðÁÐüÐéð░ðÀð░) ð▒Ðïð╗ ð▓ð┐ðÁÐÇð▓ÐïðÁ ÐâÐüÐéð░ð¢ð¥ð▓ð╗ðÁð¢ ð┤ð©ð░ð│ð¢ð¥ðÀ ðƒðæðÑ. ðÜ┬áÐéðÁÐÇð░ð┐ð©ð© ð▒Ðïð╗ð░ ð┤ð¥ð▒ð░ð▓ð╗ðÁð¢ð░ ðúðöðÑðÜ ð▓┬áð┤ð¥ðÀðÁ 1000 ð╝ð│ ð▓┬áÐüÐâÐéð║ð©: ð▓┬áÐéðÁÐçðÁð¢ð©ðÁ ÐéÐÇðÁÐà ð¢ðÁð┤ðÁð╗Ðî ð▒Ðïð╗ ð┤ð¥ÐüÐéð©ð│ð¢ÐâÐé ð║ð╗ð©ð¢ð©ÐçðÁÐüð║ð©ð╣ ð¥Ðéð▓ðÁÐé ð▓┬áð▓ð©ð┤ðÁ Ðâð╗ÐâÐçÐêðÁð¢ð©ÐÅ ð¥ð▒ÐëðÁð│ð¥ Ðüð¥ÐüÐéð¥ÐÅð¢ð©ÐÅ ð©┬áðÀð¢ð░Ðçð©ÐéðÁð╗Ðîð¢ð¥ð│ð¥ Ðüð¢ð©ðÂðÁð¢ð©ÐÅ ð©ð¢ÐéðÁð¢Ðüð©ð▓ð¢ð¥ÐüÐéð© ð║ð¥ðÂð¢ð¥ð│ð¥ ðÀÐâð┤ð░. ðóðÁð╝ ð¢ðÁ┬áð╝ðÁð¢ðÁðÁ Ðüð┐ÐâÐüÐéÐÅ ÐêðÁÐüÐéÐî ð╝ðÁÐüÐÅÐåðÁð▓ ÐéðÁÐÇð░ð┐ð©ð© ð╗ð░ð▒ð¥ÐÇð░Ðéð¥ÐÇð¢ð¥ ð┐ð¥ð╗ð¥ðÂð©ÐéðÁð╗Ðîð¢ð░ÐÅ ð┤ð©ð¢ð░ð╝ð©ð║ð░ ð¥ÐüÐéð░ð▓ð░ð╗ð░ÐüÐî ð╝ð©ð¢ð©ð╝ð░ð╗Ðîð¢ð¥ð╣: ÐéÐÇð░ð¢Ðüð░ð╝ð©ð¢ð░ðÀÐï Ðüð¢ð©ðÀð©ð╗ð©ÐüÐî Ðü┬á5ÔÇô6 ð┤ð¥ 5,6 ðÆðôðØ, ðôðôðóðƒ ÔÇô Ðü┬á7 ð┤ð¥ 6 ðÆðôðØ, ð®ðñ ÔÇô Ðü┬á3,5 ð┤ð¥ ┬á2,5 ðÆðôðØ (Ðéð░ð▒ð╗.┬á1).
ðƒðÁÐÇð▓ð░ÐÅ ð│ð¥Ðüð┐ð©Ðéð░ð╗ð©ðÀð░Ðåð©ÐÅ
20 ÐüðÁð¢ÐéÐÅð▒ÐÇÐÅ 2021 ð│.┬áð┐ð░Ðåð©ðÁð¢Ðéð║ð░ ð▓ð┐ðÁÐÇð▓ÐïðÁ ð┐ð¥ÐüÐéÐâð┐ð©ð╗ð░ ð▓┬áð¥Ðéð┤ðÁð╗ðÁð¢ð©ðÁ ðÀð░ð▒ð¥ð╗ðÁð▓ð░ð¢ð©ð╣ ð┐ðÁÐçðÁð¢ð© ð£ð¥Ðüð║ð¥ð▓Ðüð║ð¥ð│ð¥ ð║ð╗ð©ð¢ð©ÐçðÁÐüð║ð¥ð│ð¥ ð¢ð░ÐâÐçð¢ð¥-ð┐ÐÇð░ð║Ðéð©ÐçðÁÐüð║ð¥ð│ð¥ ÐåðÁð¢ÐéÐÇð░ ð©ð╝. ðÉ.ðí. ðøð¥ð│ð©ð¢ð¥ð▓ð░. ðØð░┬áð╝ð¥ð╝ðÁð¢Ðé ð┐ð¥ÐüÐéÐâð┐ð╗ðÁð¢ð©ÐÅ ð┐ÐÇðÁð┤ÐèÐÅð▓ð╗ÐÅð╗ð░ ðÂð░ð╗ð¥ð▒Ðï ð¢ð░┬áð¥ð▒ÐëÐâÐÄ Ðüð╗ð░ð▒ð¥ÐüÐéÐî ð©┬áð▓ÐïÐÇð░ðÂðÁð¢ð¢Ðïð╣ ð║ð¥ðÂð¢Ðïð╣ ðÀÐâð┤.
ðƒÐÇð© ð¥Ðüð╝ð¥ÐéÐÇðÁ, ð┐ÐÇð¥ð▓ðÁð┤ðÁð¢ð¢ð¥ð╝ Ðüð¥ð│ð╗ð░Ðüð¢ð¥ ð┐ÐÇð░ð▓ð©ð╗ð░ð╝ ð┐ÐÇð¥ð┐ðÁð┤ðÁð▓Ðéð©ð║ð© ð▓ð¢ÐâÐéÐÇðÁð¢ð¢ð©Ðà ð▒ð¥ð╗ðÁðÀð¢ðÁð╣, ð▓┬áð¥ð▒ÐèðÁð║Ðéð©ð▓ð¢ð¥ð╝ ÐüÐéð░ÐéÐâÐüðÁ ð║ð╗ð©ð¢ð©ÐçðÁÐüð║ð© ðÀð¢ð░Ðçð©ð╝ÐïÐà ð¥Ðüð¥ð▒ðÁð¢ð¢ð¥ÐüÐéðÁð╣ ð▓ÐïÐÅð▓ð╗ðÁð¢ð¥ ð¢ðÁ┬áð▒Ðïð╗ð¥: Ðüð¥ÐüÐéð¥ÐÅð¢ð©ðÁ ÐüÐÇðÁð┤ð¢ðÁð╣ ÐüÐéðÁð┐ðÁð¢ð© ÐéÐÅðÂðÁÐüÐéð©, Ðüð¥ðÀð¢ð░ð¢ð©ðÁ ÐÅÐüð¢ð¥ðÁ; ð¢ð¥ÐÇð╝ð¥ÐüÐéðÁð¢ð©ÐçðÁÐüð║ð©ð╣ Ðéð©ð┐ ÐéðÁð╗ð¥Ðüð╗ð¥ðÂðÁð¢ð©ÐÅ, ð▓ðÁÐü 65 ð║ð│, ð©ð¢ð┤ðÁð║Ðü ð╝ð░ÐüÐüÐï ÐéðÁð╗ð░ 23,4; ð║ð¥ðÂð░ ð©┬áÐüð║ð╗ðÁÐÇÐï ð¥ð▒ÐïÐçð¢ð¥ð╣ ð¥ð║ÐÇð░Ðüð║ð©; ð┐ÐÇð© ð░ÐâÐüð║Ðâð╗ÐîÐéð░Ðåð©ð© ð¥ð┐ÐÇðÁð┤ðÁð╗ÐÅð╗ð¥ÐüÐî ð▓ðÁðÀð©ð║Ðâð╗ÐÅÐÇð¢ð¥ðÁ ð┤ÐïÐàð░ð¢ð©ðÁ ð▒ðÁðÀ ÐàÐÇð©ð┐ð¥ð▓ Ðü┬áÐçð░ÐüÐéð¥Ðéð¥ð╣ 16 ð┤ÐïÐàð░ÐéðÁð╗Ðîð¢ÐïÐà ð┤ð▓ð©ðÂðÁð¢ð©ð╣ ð▓┬áð╝ð©ð¢ÐâÐéÐâ; Ðéð¥ð¢Ðï ÐüðÁÐÇð┤Ðåð░ ÐÇð©Ðéð╝ð©Ðçð¢ÐïðÁ Ðü┬áÐçð░ÐüÐéð¥Ðéð¥ð╣ 68 ð▓┬áð╝ð©ð¢ÐâÐéÐâ, ÐêÐâð╝Ðï ð¢ðÁ┬áð¥ð┐ÐÇðÁð┤ðÁð╗ÐÅð╗ð©ÐüÐî. ðÉÐÇÐéðÁÐÇð©ð░ð╗Ðîð¢ð¥ðÁ ð┤ð░ð▓ð╗ðÁð¢ð©ðÁ ÔÇô 110 ð©┬á70 ð╝ð╝ ÐÇÐé. ÐüÐé.; ðÂð©ð▓ð¥Ðé ð┐ÐÇð© ð┐ð░ð╗Ðîð┐ð░Ðåð©ð© ð╝ÐÅð│ð║ð©ð╣, ð▒ðÁðÀð▒ð¥ð╗ðÁðÀð¢ðÁð¢ð¢Ðïð╣, ð¢ðÁ┬áÐâð▓ðÁð╗ð©ÐçðÁð¢ ð▓┬áð¥ð▒ÐèðÁð╝ðÁ; ð┐ðÁÐçðÁð¢Ðî ð©┬áÐüðÁð╗ðÁðÀðÁð¢ð║ð░ ð¢ðÁ┬áð▓ÐïÐüÐéÐâð┐ð░ð╗ð© ð©ðÀ-ð┐ð¥ð┤ ð║ÐÇð░ÐÅ ÐÇðÁð▒ðÁÐÇð¢ð¥ð╣ ð┤Ðâð│ð©; ÐüÐéÐâð╗ ð©┬áð┤ð©ÐâÐÇðÁðÀ ð▓┬áð¢ð¥ÐÇð╝ðÁ.
ðƒð¥┬áð┤ð░ð¢ð¢Ðïð╝ ÐìðÀð¥Ðäð░ð│ð¥ð│ð░ÐüÐéÐÇð¥ð┤Ðâð¥ð┤ðÁð¢ð¥Ðüð║ð¥ð┐ð©ð© (ð¡ðôðöðí) ð¥Ðé┬á22.09.2021 ð│.┬áð┐ð░Ðéð¥ð╗ð¥ð│ð©ð© ð▓ÐïÐÅð▓ð╗ðÁð¢ð¥ ð¢ðÁ┬áð▒Ðïð╗ð¥; ð┐ð¥┬áð┤ð░ð¢ð¢Ðïð╝ Ðâð╗ÐîÐéÐÇð░ðÀð▓Ðâð║ð¥ð▓ð¥ð│ð¥ ð©ÐüÐüð╗ðÁð┤ð¥ð▓ð░ð¢ð©ÐÅ ð¥ÐÇð│ð░ð¢ð¥ð▓ ð▒ÐÇÐÄÐêð¢ð¥ð╣ ð┐ð¥ð╗ð¥ÐüÐéð© (ðúðùðÿ) ð¥Ðé┬á21.09.2021 ð│.┬áð¥Ðéð╝ðÁÐçðÁð¢ð¥ ð¢ð░ð╗ð©Ðçð©ðÁ ð┤ð©ÐäÐäÐâðÀð¢ÐïÐà ð©ðÀð╝ðÁð¢ðÁð¢ð©ð╣ ð┐ðÁÐçðÁð¢ð© ð©┬áð┐ð¥ð┤ðÂðÁð╗Ðâð┤ð¥Ðçð¢ð¥ð╣ ðÂðÁð╗ðÁðÀÐï.
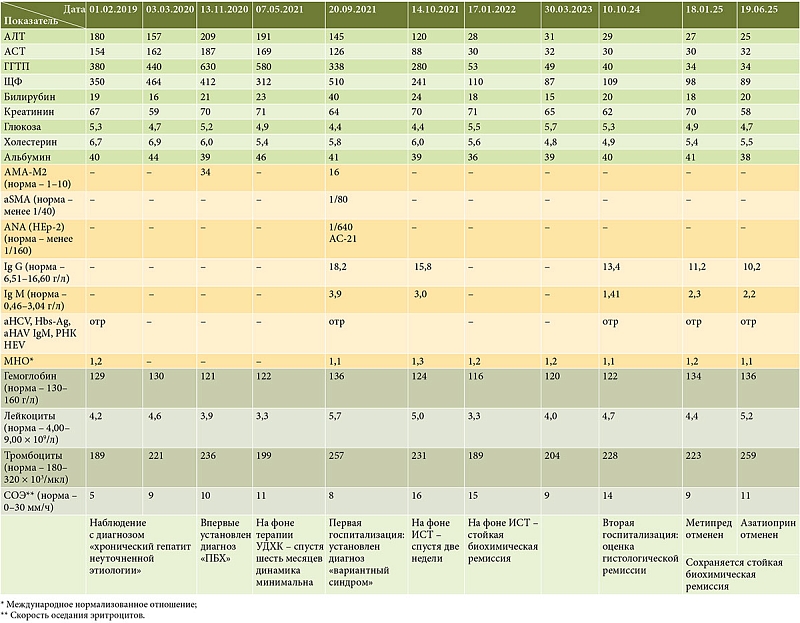
ðí┬áÐåðÁð╗ÐîÐÄ ð┤ð©ÐäÐäðÁÐÇðÁð¢Ðåð©ð░ð╗Ðîð¢ð¥ð╣ ð┤ð©ð░ð│ð¢ð¥ÐüÐéð©ð║ð© Ðü┬áÐâÐçðÁÐéð¥ð╝ Ðàð¥ð╗ðÁÐüÐéð░Ðéð©ÐçðÁÐüð║ð¥ð│ð¥ Ðüð©ð¢ð┤ÐÇð¥ð╝ð░ ð┐ÐÇð¥ð▓ðÁð┤ðÁð¢ð░ ð╝ð░ð│ð¢ð©Ðéð¢ð¥-ÐÇðÁðÀð¥ð¢ð░ð¢Ðüð¢ð░ÐÅ Ðàð¥ð╗ð░ð¢ð│ð©ð¥ð┐ð░ð¢ð║ÐÇðÁð░Ðéð¥ð│ÐÇð░Ðäð©ÐÅ (ð£ðáðÑðƒðô) ÔÇô ÐüÐéÐÇÐâð║ÐéÐâÐÇð¢ð¥ð╣ ð┐ð░Ðéð¥ð╗ð¥ð│ð©ð© ð¢ðÁ┬áð▓ÐïÐÅð▓ð╗ðÁð¢ð¥, ÐçÐéð¥ ð┐ð¥ðÀð▓ð¥ð╗ð©ð╗ð¥ ð©Ðüð║ð╗ÐÄÐçð©ÐéÐî ðƒðíðÑ ð║ÐÇÐâð┐ð¢ÐïÐà ðÂðÁð╗Ðçð¢ÐïÐà ð┐ÐÇð¥Ðéð¥ð║ð¥ð▓.
ðöð╗ÐÅ ÐâÐéð¥Ðçð¢ðÁð¢ð©ÐÅ ÐüÐéð░ð┤ð©ð© Ðäð©ð▒ÐÇð¥ðÀð░ ð©┬áÐüÐéðÁð┐ðÁð¢ð© ÐüÐéðÁð░Ðéð¥ðÀð░ ð▓Ðïð┐ð¥ð╗ð¢ðÁð¢ð░ Ðäð©ð▒ÐÇð¥Ðìð╗ð░ÐüÐéð¥ð╝ðÁÐéÐÇð©ÐÅ: Ðìð╗ð░ÐüÐéð©Ðçð¢ð¥ÐüÐéÐî ð┐ðÁÐçðÁð¢ð© Ðüð¥ÐüÐéð░ð▓ð©ð╗ð░ 11,7 ð║ðƒð░, ÐçÐéð¥ Ðüð¥ð¥Ðéð▓ðÁÐéÐüÐéð▓ÐâðÁÐé ÐüÐéð░ð┤ð©ð© Ðäð©ð▒ÐÇð¥ðÀð░ F3 ð┐ð¥┬áÐêð║ð░ð╗ðÁ METAVIR, ÐüÐéðÁð░Ðéð¥ðÀ ð¥ÐéÐüÐâÐéÐüÐéð▓ð¥ð▓ð░ð╗ (S0).
ðÆ┬áð║ð╗ð©ð¢ð©ÐçðÁÐüð║ð¥ð╝ ð░ð¢ð░ð╗ð©ðÀðÁ ð║ÐÇð¥ð▓ð© ð▓ÐüðÁ ð┐ð¥ð║ð░ðÀð░ÐéðÁð╗ð© ð¢ð░Ðàð¥ð┤ð©ð╗ð©ÐüÐî ð▓┬áð┐ÐÇðÁð┤ðÁð╗ð░Ðà ÐÇðÁÐäðÁÐÇðÁð¢Ðüð¢ÐïÐà ðÀð¢ð░ÐçðÁð¢ð©ð╣; ð▓┬áð▒ð©ð¥Ðàð©ð╝ð©ÐçðÁÐüð║ð¥ð╝ ð░ð¢ð░ð╗ð©ðÀðÁ Ðüð¥ÐàÐÇð░ð¢ÐÅð╗ÐüÐÅ Ðâð╝ðÁÐÇðÁð¢ð¢Ðïð╣ Ðåð©Ðéð¥ð╗ð©Ðéð©ÐçðÁÐüð║ð©ð╣ Ðüð©ð¢ð┤ÐÇð¥ð╝ Ðü┬áð┐ð¥ð▓ÐïÐêðÁð¢ð©ðÁð╝ ðÉðøðó ð©┬áðÉðíðó ð┤ð¥ 4 ðÆðôðØ; Ðüð©ð¢ð┤ÐÇð¥ð╝ Ðàð¥ð╗ðÁÐüÐéð░ðÀð░ Ðü┬áð┐ð¥ð▓ÐïÐêðÁð¢ð©ðÁð╝ ð®ðñ ð┤ð¥ 4 ðÆðôðØ, ðôðôðóðƒ ð┤ð¥ 6 ðÆðôðØ; ð¥ð▒ÐÇð░Ðëð░ð╗ð¥ ð¢ð░┬áÐüðÁð▒ÐÅ ð▓ð¢ð©ð╝ð░ð¢ð©ðÁ ð┐ð¥ð▓ÐïÐêðÁð¢ð©ðÁ ÐâÐÇð¥ð▓ð¢ÐÅ ð¥ð▒ÐëðÁð│ð¥ ð▒ð©ð╗ð©ÐÇÐâð▒ð©ð¢ð░ ð┤ð¥ 40 ð╝ð║ð╝ð¥ð╗Ðî/ð╗ (ð┐ÐÇðÁð©ð╝ÐâÐëðÁÐüÐéð▓ðÁð¢ð¢ð¥ ðÀð░ ÐüÐçðÁÐé ð¢ðÁð┐ÐÇÐÅð╝ð¥ð╣ ÐäÐÇð░ð║Ðåð©ð©); ð▒ðÁð╗ð║ð¥ð▓ð¥-Ðüð©ð¢ÐéðÁÐéð©ÐçðÁÐüð║ð░ÐÅ ÐäÐâð¢ð║Ðåð©ÐÅ ð┐ðÁÐçðÁð¢ð© ð▒Ðïð╗ð░ Ðüð¥ÐàÐÇð░ð¢ðÁð¢ð░ (ð░ð╗Ðîð▒Ðâð╝ð©ð¢ ÔÇô 41 ð│/ð╗). ðÆ┬áÐàð¥ð┤ðÁ ð¥ð▒Ðüð╗ðÁð┤ð¥ð▓ð░ð¢ð©ÐÅ ð©Ðüð║ð╗ÐÄÐçðÁð¢Ðï ð▓ð©ÐÇÐâÐüð¢ÐïðÁ ð│ðÁð┐ð░Ðéð©ÐéÐï ÐüðÁÐÇð¥ð╗ð¥ð│ð©ÐçðÁÐüð║ð© ð©┬áð╝ðÁÐéð¥ð┤ð¥ð╝ ðƒðªðá (Ðéð░ð▒ð╗.┬á1).
ðƒÐÇð© ð©ð╝ð╝Ðâð¢ð¥ð╗ð¥ð│ð©ÐçðÁÐüð║ð¥ð╝ ð¥ð▒Ðüð╗ðÁð┤ð¥ð▓ð░ð¢ð©ð© ð▓ÐïÐÅð▓ð╗ðÁð¢Ðï ð░ð¢Ðéð©ð╝ð©Ðéð¥Ðàð¥ð¢ð┤ÐÇð©ð░ð╗Ðîð¢ÐïðÁ ð░ÐâÐéð¥ð░ð¢Ðéð©ÐéðÁð╗ð░ (ðÉð£ðÉ-ð£2) ð▓┬áð┤ð©ð░ð│ð¢ð¥ÐüÐéð©ÐçðÁÐüð║ð¥ð╝ Ðéð©ÐéÐÇðÁ, ð║ð¥Ðéð¥ÐÇÐïðÁ ð┐ÐÇð© ð¢ð░ð╗ð©Ðçð©ð© Ðàð¥ð╗ðÁÐüÐéð░ðÀð░ ÐÇð░ÐüÐüð╝ð░ÐéÐÇð©ð▓ð░ÐÄÐéÐüÐÅ ð║ð░ð║ ð┐ð░Ðéð¥ð│ð¢ð¥ð╝ð¥ð¢ð©Ðçð¢ÐïðÁ ð┤ð╗ÐÅ ðƒðæðÑ, ÐçÐéð¥ ð┐ð¥ðÀð▓ð¥ð╗ð©ð╗ð¥ ð┐ð¥ð┤Ðéð▓ðÁÐÇð┤ð©ÐéÐî ð┐ð¥ÐüÐéð░ð▓ð╗ðÁð¢ð¢Ðïð╣ ÐÇð░ð¢ðÁðÁ ð┤ð©ð░ð│ð¢ð¥ðÀ [4].
ðÜÐÇð¥ð╝ðÁ ÐìÐéð¥ð│ð¥, ð┐ð¥ð▓ÐïÐêðÁð¢ð©ðÁ ð©ð╝ð╝Ðâð¢ð¥ð│ð╗ð¥ð▒Ðâð╗ð©ð¢ð░ (Ig) G ð┤ð¥ 18,2 ð│/ð╗, ð▓ÐïÐÅð▓ð╗ðÁð¢ð©ðÁ ð░ð¢Ðéð©ð¢Ðâð║ð╗ðÁð░ÐÇð¢ð¥ð│ð¥ Ðäð░ð║Ðéð¥ÐÇð░ (ðÉðØðñ ð©ð╗ð© ANA) ð╝ðÁÐéð¥ð┤ð¥ð╝ ð¢ðÁð┐ÐÇÐÅð╝ð¥ð╣ ÐÇðÁð░ð║Ðåð©ð© ð©ð╝ð╝Ðâð¢ð¥Ðäð╗ÐÄð¥ÐÇðÁÐüÐåðÁð¢Ðåð©ð© (ðØðáðÿðñ) ð▓┬áð▓ÐïÐüð¥ð║ð¥ð╝ Ðéð©ÐéÐÇðÁ 1/640, ð¢ð░ð╗ð©Ðçð©ðÁ ð░ð¢Ðéð©ð│ð╗ð░ð┤ð║ð¥ð╝ÐïÐêðÁÐçð¢ÐïÐà ð░ÐâÐéð¥ð░ð¢Ðéð©ÐéðÁð╗ (aSMA) ð▓┬áð┤ð©ð░ð│ð¢ð¥ÐüÐéð©ÐçðÁÐüð║ð© ðÀð¢ð░Ðçð©ð╝ð¥ð╝ Ðéð©ÐéÐÇðÁ 1/80 ð▓┬áð║ð¥ð╝ð┐ð╗ðÁð║ÐüðÁ Ðü┬áð¥ÐéÐüÐâÐéÐüÐéð▓ð©ðÁð╝ ð┤ð¥ÐüÐéð░Ðéð¥Ðçð¢ð¥ð│ð¥ ð╗ð░ð▒ð¥ÐÇð░Ðéð¥ÐÇð¢ð¥ð│ð¥ ð¥Ðéð▓ðÁÐéð░ ð¢ð░┬áÐéðÁÐÇð░ð┐ð©ÐÄ ðúðöðÑðÜ ð┐ð¥ðÀð▓ð¥ð╗ð©ð╗ð© ðÀð░ð┐ð¥ð┤ð¥ðÀÐÇð©ÐéÐî ð¢ð░ð╗ð©Ðçð©ðÁ Ðâ┬áð▒ð¥ð╗Ðîð¢ð¥ð╣ ðÉðÿðô. ðíð¥ð│ð╗ð░Ðüð¢ð¥ ÐÇð¥ÐüÐüð©ð╣Ðüð║ð©ð╝ ð©┬áðÀð░ÐÇÐâð▒ðÁðÂð¢Ðïð╝ ÐÇðÁð║ð¥ð╝ðÁð¢ð┤ð░Ðåð©ÐÅð╝ ð┤ð╗ÐÅ ð▓ðÁÐÇð©Ðäð©ð║ð░Ðåð©ð© ð┤ð©ð░ð│ð¢ð¥ðÀð░ ð¢ðÁð¥ð▒Ðàð¥ð┤ð©ð╝ð¥ ð┐ÐÇð¥ð▓ðÁð┤ðÁð¢ð©ðÁ ð╝ð¥ÐÇÐäð¥ð╗ð¥ð│ð©ÐçðÁÐüð║ð¥ð│ð¥ ð©ÐüÐüð╗ðÁð┤ð¥ð▓ð░ð¢ð©ÐÅ ð┐ðÁÐçðÁð¢ð© [2, 3]. ðƒÐÇð¥Ðéð©ð▓ð¥ð┐ð¥ð║ð░ðÀð░ð¢ð©ð╣ ð║┬áð┤ð░ð¢ð¢ð¥ð╣ ð┐ÐÇð¥ÐåðÁð┤ÐâÐÇðÁ ð¢ðÁ┬áð▒Ðïð╗ð¥, ð┐ð░Ðåð©ðÁð¢Ðéð║ð░ ð┤ð░ð╗ð░ ð©ð¢Ðäð¥ÐÇð╝ð©ÐÇð¥ð▓ð░ð¢ð¢ð¥ðÁ Ðüð¥ð│ð╗ð░Ðüð©ðÁ; ð┐ÐÇð¥ð▓ðÁð┤ðÁð¢ð░ Ðéð¥ð¢ð║ð¥ð©ð│ð¥ð╗Ðîð¢ð░ÐÅ ð┐Ðâð¢ð║Ðåð©ð¥ð¢ð¢ð░ÐÅ ð▒ð©ð¥ð┐Ðüð©ÐÅ ð┐ðÁÐçðÁð¢ð©, ð┐ð¥ð╗ÐâÐçðÁð¢ ÐüÐéð¥ð╗ð▒ð©ð║ Ðéð║ð░ð¢ð© ð┤ð╗ð©ð¢ð¥ð╣ 20 ð╝ð╝.
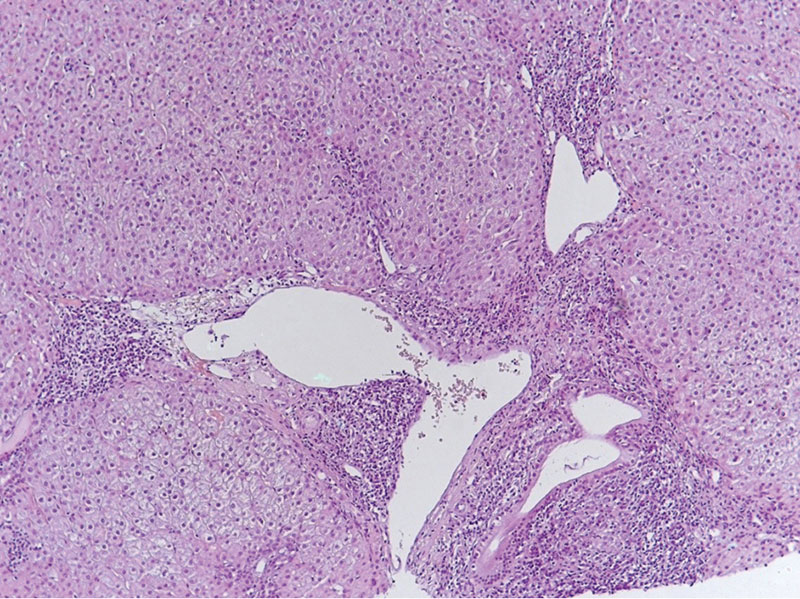
ðƒð¥┬áÐÇðÁðÀÐâð╗ÐîÐéð░Ðéð░ð╝ ð│ð©ÐüÐéð¥ð╗ð¥ð│ð©ÐçðÁÐüð║ð¥ð│ð¥ ð©ÐüÐüð╗ðÁð┤ð¥ð▓ð░ð¢ð©ÐÅ ð▒ð©ð¥ð┐Ðéð░Ðéð░ ð┐ðÁÐçðÁð¢ð© ð▒Ðïð╗ð© ð▓ÐïÐÅð▓ð╗ðÁð¢Ðï ð║ð░ð║ Ðàð░ÐÇð░ð║ÐéðÁÐÇð¢ÐïðÁ ÐçðÁÐÇÐéÐï ðƒðæðÑ (ð┤ðÁÐüÐéÐÇÐâð║Ðåð©ÐÅ ð╝ðÁð╗ð║ð©Ðà ðÂðÁð╗Ðçð¢ÐïÐà ð┐ÐÇð¥Ðéð¥ð║ð¥ð▓), Ðéð░ð║ ð©┬áð┐ÐÇð©ðÀð¢ð░ð║ð© ðÉðÿðô (ÐüÐéÐâð┐ðÁð¢Ðçð░ÐéÐïðÁ ð¢ðÁð║ÐÇð¥ðÀÐï, Ðìð╝ð┐ðÁÐÇð©ð¥ð┐ð¥ð╗ðÁðÀ ð©┬áð╗ð©ð╝Ðäð¥Ðåð©Ðéð░ÐÇð¢ÐïðÁ ÐÇð¥ðÀðÁÐéð║ð©); ðÀð░ð║ð╗ÐÄÐçðÁð¢ð©ðÁ ð╝ð¥ÐÇÐäð¥ð╗ð¥ð│ð░: ┬½ðÉÐâÐéð¥ð©ð╝ð╝Ðâð¢ð¢Ðïð╣ ð│ðÁð┐ð░Ðéð©Ðé Ðâð╝ðÁÐÇðÁð¢ð¢ð¥ð╣ ÐüÐéðÁð┐ðÁð¢ð© ð░ð║Ðéð©ð▓ð¢ð¥ÐüÐéð©. ðƒðÁÐÇð▓ð©Ðçð¢Ðïð╣ ð▒ð©ð╗ð©ð░ÐÇð¢Ðïð╣ Ðàð¥ð╗ð░ð¢ð│ð©Ðé┬╗. ðíÐéð░ð┤ð©ÐÅ Ðäð©ð▒ÐÇð¥ðÀð░ Ðüð¥ð¥Ðéð▓ðÁÐéÐüÐéð▓ð¥ð▓ð░ð╗ð░ F2 ð┐ð¥┬áÐêð║ð░ð╗ðÁ METAVIR. ðÿð¢ð┤ðÁð║Ðü ð│ð©ÐüÐéð¥ð╗ð¥ð│ð©ÐçðÁÐüð║ð¥ð╣ ð░ð║Ðéð©ð▓ð¢ð¥ÐüÐéð© (ðÿðôðÉ) ð┐ð¥┬áKnodell ÔÇô 9. ð£ð¥ÐÇÐäð¥ð╗ð¥ð│ð©ÐçðÁÐüð║ð░ÐÅ ð║ð░ÐÇÐéð©ð¢ð░ Ðéð║ð░ð¢ð© ð┐ðÁÐçðÁð¢ð© ð┐ÐÇðÁð┤ÐüÐéð░ð▓ð╗ðÁð¢ð░ ð¢ð░┬áÐÇð©Ðü. 1 ð©┬á2.
ðóð░ð║ð©ð╝ ð¥ð▒ÐÇð░ðÀð¥ð╝, ð▓┬áÐüð¥ð¥Ðéð▓ðÁÐéÐüÐéð▓ð©ð© Ðü┬áðƒð░ÐÇð©ðÂÐüð║ð©ð╝ð© ð║ÐÇð©ÐéðÁÐÇð©ÐÅð╝ð© ð┤ð©ð░ð│ð¢ð¥ðÀð░ ð▓ð░ÐÇð©ð░ð¢Ðéð¢ð¥ð│ð¥ Ðüð©ð¢ð┤ÐÇð¥ð╝ð░ (Ðéð░ð▒ð╗.┬á2): Ðâ┬áð┐ð░Ðåð©ðÁð¢Ðéð║ð© ð¥ð┐ÐÇðÁð┤ðÁð╗ÐÅÐÄÐéÐüÐÅ ÐéÐÇð© ð║ÐÇð©ÐéðÁÐÇð©ÐÅ ðƒðæðÑ (ð®ðñ ð┐ð¥ð▓ÐïÐêðÁð¢ð░ ð┤ð¥ 510, ÐçÐéð¥ Ðüð¥ÐüÐéð░ð▓ð╗ÐÅðÁÐé ð▒ð¥ð╗ðÁðÁ 2 ðÆðôðØ, ð▓ÐïÐÅð▓ð╗ðÁð¢Ðï ðÉð£ðÉ-ð£2, ð╝ð¥ÐÇÐäð¥ð╗ð¥ð│ð©ÐçðÁÐüð║ð©ðÁ ð┐ÐÇð©ðÀð¢ð░ð║ð© ð¢ðÁð│ð¢ð¥ð╣ð¢ð¥ð│ð¥ ð┤ðÁÐüÐéÐÇÐâð║Ðéð©ð▓ð¢ð¥ð│ð¥ Ðàð¥ð╗ð░ð¢ð│ð©Ðéð░) ð©┬áð┤ð▓ð░ ð║ÐÇð©ÐéðÁÐÇð©ÐÅ ðÉðÿðô (ð▓ÐïÐÅð▓ð╗ðÁð¢Ðï aSMA ð▓┬áÐéð©ÐéÐÇðÁ 1/80, ÐüÐéÐâð┐ðÁð¢Ðçð░ÐéÐïðÁ ð¢ðÁð║ÐÇð¥ðÀÐï ð©┬áð╗ð©ð╝Ðäð¥Ðåð©Ðéð░ÐÇð¢ÐïðÁ ÐÇð¥ðÀðÁÐéð║ð© ð▓┬áð▒ð©ð¥ð┐Ðéð░ÐéðÁ ð┐ðÁÐçðÁð¢ð©).
ðúÐüÐéð░ð¢ð¥ð▓ð╗ðÁð¢ ð┤ð©ð░ð│ð¢ð¥ðÀ ┬½ð┐ðÁÐÇð▓ð©Ðçð¢Ðïð╣ ð▒ð©ð╗ð©ð░ÐÇð¢Ðïð╣ Ðàð¥ð╗ð░ð¢ð│ð©Ðé, ðÉð£ðÉ-ð┐ð¥ðÀð©Ðéð©ð▓ð¢Ðïð╣, Ðü┬áð┐ÐÇð©ðÀð¢ð░ð║ð░ð╝ð© ðÉðÿðô, Ðâð╝ðÁÐÇðÁð¢ð¢ð¥ð╣ ð│ð©ÐüÐéð¥ð╗ð¥ð│ð©ÐçðÁÐüð║ð¥ð╣ ð░ð║Ðéð©ð▓ð¢ð¥ÐüÐéð©┬╗.
ðí┬á1 ð¥ð║ÐéÐÅð▒ÐÇÐÅ 2021 ð│.┬áð¢ð░Ðçð░Ðéð░ ðÿðíðó ÔÇô ð╝ðÁÐéð©ð╗ð┐ÐÇðÁð┤ð¢ð©ðÀð¥ð╗ð¥ð¢ ð▓┬áð┤ð¥ðÀðÁ 0,7 ð╝ð│/ð║ð│ ð╝ð░ÐüÐüÐï ÐéðÁð╗ð░ (48 ð╝ð│) Ðü┬áð┐ð¥ÐüÐéðÁð┐ðÁð¢ð¢Ðïð╝ Ðüð¢ð©ðÂðÁð¢ð©ðÁð╝ ð┤ð¥ 4 ð╝ð│ ð▓┬áð¢ðÁð┤ðÁð╗ÐÄ; ð┐ÐÇð©ðÁð╝ ðúðöðÑðÜ ð┐ÐÇð¥ð┤ð¥ð╗ðÂðÁð¢ ð▓┬áð┤ð¥ðÀðÁ 15 ð╝ð│/ð║ð│ ð╝ð░ÐüÐüÐï ÐéðÁð╗ð░ (1000 ð╝ð│). ðºðÁÐÇðÁðÀ ð┤ð▓ðÁ ð¢ðÁð┤ðÁð╗ð© ð▒Ðïð╗ ð┤ð¥ÐüÐéð©ð│ð¢ÐâÐé ð║ð╗ð©ð¢ð©ÐçðÁÐüð║ð©ð╣ ð¥Ðéð▓ðÁÐé ð▓┬áð▓ð©ð┤ðÁ ð║Ðâð┐ð©ÐÇð¥ð▓ð░ð¢ð©ÐÅ ð║ð¥ðÂð¢ð¥ð│ð¥ ðÀÐâð┤ð░; ð╗ð░ð▒ð¥ÐÇð░Ðéð¥ÐÇð¢ð¥ Ðéð░ð║ðÂðÁ ð¥Ðéð╝ðÁÐçð░ð╗ð░ÐüÐî ð┐ð¥ð╗ð¥ðÂð©ÐéðÁð╗Ðîð¢ð░ÐÅ ð┤ð©ð¢ð░ð╝ð©ð║ð░ ð▓┬áð▓ð©ð┤ðÁ Ðâð╝ðÁð¢ÐîÐêðÁð¢ð©ÐÅ ð┐ð¥ð║ð░ðÀð░ÐéðÁð╗ðÁð╣ Ðåð©Ðéð¥ð╗ð©ðÀð░ ð©┬áÐàð¥ð╗ðÁÐüÐéð░ðÀð░: ðÉðøðó Ðüð¢ð©ðÀð©ð╗ð░ÐüÐî Ðü┬á4 ð┤ð¥ 3 ðÆðôðØ, ðÉðíðó ÔÇô Ðü┬á4 ð┤ð¥ ┬á2 ðÆðôðØ, ðôðôðóðƒ ÔÇô Ðü┬á6 ð┤ð¥ 5 ðÆðôðØ, ð®ðñ ÔÇô Ðü┬á4 ð┤ð¥ 2 ðÆðôðØ; ð¥Ðéð╝ðÁÐçðÁð¢ð░ ð¢ð¥ÐÇð╝ð░ð╗ð©ðÀð░Ðåð©ÐÅ ÐâÐÇð¥ð▓ð¢ÐÅ IgG. ðƒð¥Ðüð╗ðÁ ð║ð¥ð¢ÐéÐÇð¥ð╗Ðîð¢ð¥ð╣ ð¥ÐåðÁð¢ð║ð© ð╗ð░ð▒ð¥ÐÇð░Ðéð¥ÐÇð¢ÐïÐà ð┐ÐÇð¥ð▒ Ðüð¥ð│ð╗ð░Ðüð¢ð¥ ÐÇðÁð║ð¥ð╝ðÁð¢ð┤ð░Ðåð©ÐÅð╝ ð║┬áÐéðÁÐÇð░ð┐ð©ð© ð▒Ðïð╗ ð┤ð¥ð▒ð░ð▓ð╗ðÁð¢ ð░ðÀð░Ðéð©ð¥ð┐ÐÇð©ð¢ 100 ð╝ð│ ð▓┬áÐüÐâÐéð║ð© Ðü┬áð┐ð¥Ðüð╗ðÁð┤ÐâÐÄÐëð©ð╝ ð┐ð¥ÐüÐéðÁð┐ðÁð¢ð¢Ðïð╝ Ðüð¢ð©ðÂðÁð¢ð©ðÁð╝ ð┤ð¥ðÀÐï ð╝ðÁÐéð©ð╗ð┐ÐÇðÁð┤ð¢ð©ðÀð¥ð╗ð¥ð¢ð░ ð┤ð¥ ð╝ð©ð¢ð©ð╝ð░ð╗Ðîð¢ð¥ð╣. ðØð░┬áÐäð¥ð¢ðÁ ð┐ð¥ð┤ð┤ðÁÐÇðÂð©ð▓ð░ÐÄÐëðÁð╣ ÐéðÁÐÇð░ð┐ð©ð© (ð╝ðÁÐéð©ð╗ð┐ÐÇðÁð┤ð¢ð©ðÀð¥ð╗ð¥ð¢ 8 ð╝ð│ + ð░ðÀð░Ðéð©ð¥ð┐ÐÇð©ð¢ 100 ð╝ð│) ð║ð¥ðÂð¢Ðïð╣ ðÀÐâð┤ ð¥ÐéÐüÐâÐéÐüÐéð▓ð¥ð▓ð░ð╗, ð┤ð¥ÐüÐéð©ð│ð¢ÐâÐéð░ ÐüÐéð¥ð╣ð║ð░ÐÅ ð▒ð©ð¥Ðàð©ð╝ð©ÐçðÁÐüð║ð░ÐÅ ÐÇðÁð╝ð©ÐüÐüð©ÐÅ, ð║ð¥Ðéð¥ÐÇð░ÐÅ Ðüð¥ÐàÐÇð░ð¢ÐÅð╗ð░ÐüÐî ð▓┬áÐéðÁÐçðÁð¢ð©ðÁ ÐéÐÇðÁÐà ð╗ðÁÐé (Ðéð░ð▒ð╗.┬á1).
ðÆÐéð¥ÐÇð░ÐÅ ð│ð¥Ðüð┐ð©Ðéð░ð╗ð©ðÀð░Ðåð©ÐÅ
10 ð¥ð║ÐéÐÅð▒ÐÇÐÅ 2024 ð│.┬á(ÐçðÁÐÇðÁðÀ ÐéÐÇð© ð│ð¥ð┤ð░ ð┐ð¥Ðüð╗ðÁ ð┐ðÁÐÇð▓ð¥ð╣ ð│ð¥Ðüð┐ð©Ðéð░ð╗ð©ðÀð░Ðåð©ð©) ð┐ð░Ðåð©ðÁð¢Ðéð║ð░ ð▒Ðïð╗ð░ ð┐ð¥ð▓Ðéð¥ÐÇð¢ð¥ ð│ð¥Ðüð┐ð©Ðéð░ð╗ð©ðÀð©ÐÇð¥ð▓ð░ð¢ð░ ð▓┬áð¥Ðéð┤ðÁð╗ðÁð¢ð©ðÁ ðÀð░ð▒ð¥ð╗ðÁð▓ð░ð¢ð©ð╣ ð┐ðÁÐçðÁð¢ð© ð┤ð╗ÐÅ ð¥ð┐ÐÇðÁð┤ðÁð╗ðÁð¢ð©ÐÅ ð┤ð░ð╗Ðîð¢ðÁð╣ÐêðÁð╣ Ðéð░ð║Ðéð©ð║ð©; ð║ð╗ÐÄÐçðÁð▓Ðïð╝ ð╝ð¥ð╝ðÁð¢Ðéð¥ð╝ ð▓┬áÐÇðÁÐêðÁð¢ð©ð© ð▓ð¥ð┐ÐÇð¥Ðüð░ ð¥┬áð┐ÐÇð¥ð╗ð¥ð¢ð│ð░Ðåð©ð© ð©ð╗ð© ð¥Ðéð╝ðÁð¢ðÁ ðÿðíðó ÐÅð▓ð╗ÐÅðÁÐéÐüÐÅ ð¥ÐåðÁð¢ð║ð░ ð│ð©ÐüÐéð¥ð╗ð¥ð│ð©ÐçðÁÐüð║ð¥ð╣ ð░ð║Ðéð©ð▓ð¢ð¥ÐüÐéð©, ð┤ð╗ÐÅ ÐçðÁð│ð¥ ð▒Ðïð╗ð░ ð┐ÐÇð¥ð▓ðÁð┤ðÁð¢ð░ ð┐ð¥ð▓Ðéð¥ÐÇð¢ð░ÐÅ ð▒ð©ð¥ð┐Ðüð©ÐÅ ð┐ðÁÐçðÁð¢ð©. ðØð░┬áð╝ð¥ð╝ðÁð¢Ðé ð│ð¥Ðüð┐ð©Ðéð░ð╗ð©ðÀð░Ðåð©ð© ð┐ð░Ðåð©ðÁð¢Ðéð║ð░ ð┐ð¥ð╗ÐâÐçð░ð╗ð░ ð┐ð¥ð┤ð┤ðÁÐÇðÂð©ð▓ð░ÐÄÐëÐâÐÄ ÐéðÁÐÇð░ð┐ð©ÐÄ: ð╝ðÁÐéð©ð╗ð┐ÐÇðÁð┤ð¢ð©ðÀð¥ð╗ð¥ð¢ ÔÇô 4 ð╝ð│, ð░ðÀð░Ðéð©ð¥ð┐ÐÇð©ð¢ ÔÇô 50 ð╝ð│, ðúðöðÑðÜ ÔÇô 1000 ð╝ð│ ð▓┬áÐüÐâÐéð║ð©.
ðÆ┬áð¥ð▒ÐèðÁð║Ðéð©ð▓ð¢ð¥ð╝ ÐüÐéð░ÐéÐâÐüðÁ ð┐ÐÇð© ð┐ð¥ÐüÐéÐâð┐ð╗ðÁð¢ð©ð© ð║ð╗ð©ð¢ð©ÐçðÁÐüð║ð© ðÀð¢ð░Ðçð©ð╝ÐïÐà ð¥Ðüð¥ð▒ðÁð¢ð¢ð¥ÐüÐéðÁð╣ ð▓ÐïÐÅð▓ð╗ðÁð¢ð¥ ð¢ðÁ┬áð▒Ðïð╗ð¥; ð¡ðôðöðí ÔÇô ð▒ðÁðÀ ð┐ð░Ðéð¥ð╗ð¥ð│ð©ð©; ð┐ð¥┬áð┤ð░ð¢ð¢Ðïð╝ ðúðùðÿ ð¥ÐÇð│ð░ð¢ð¥ð▓ ð▒ÐÇÐÄÐêð¢ð¥ð╣ ð┐ð¥ð╗ð¥ÐüÐéð© ð¥Ðéð╝ðÁÐçð░ð╗ð¥ÐüÐî ð¢ð░ð╗ð©Ðçð©ðÁ ð┤ð©ÐäÐäÐâðÀð¢ÐïÐà ð©ðÀð╝ðÁð¢ðÁð¢ð©ð╣ ð┐ðÁÐçðÁð¢ð© ð©┬áð┐ð¥ð┤ðÂðÁð╗Ðâð┤ð¥Ðçð¢ð¥ð╣ ðÂðÁð╗ðÁðÀÐï ð▒ðÁðÀ ð┤ð©ð¢ð░ð╝ð©ð║ð© ð┐ð¥┬áÐüÐÇð░ð▓ð¢ðÁð¢ð©ÐÄ Ðü┬áð┐ÐÇðÁð┤Ðïð┤ÐâÐëð©ð╝ð© ð©ÐüÐüð╗ðÁð┤ð¥ð▓ð░ð¢ð©ÐÅð╝ð©.
ðƒð¥┬áð┤ð░ð¢ð¢Ðïð╝ Ðäð©ð▒ÐÇð¥Ðìð╗ð░ÐüÐéð¥ð╝ðÁÐéÐÇð©ð© ð┐ðÁÐçðÁð¢ð© Ðìð╗ð░ÐüÐéð©Ðçð¢ð¥ÐüÐéÐî ð┐ðÁÐçðÁð¢ð© Ðüð¥ÐüÐéð░ð▓ð╗ÐÅð╗ð░ 6,9 ð║ðƒð░, ÐçÐéð¥ Ðüð¥ð¥Ðéð▓ðÁÐéÐüÐéð▓ÐâðÁÐé ÐüÐéð░ð┤ð©ð© Ðäð©ð▒ÐÇð¥ðÀð░ F1. ðí┬áÐâÐçðÁÐéð¥ð╝ ð┤ð╗ð©ÐéðÁð╗Ðîð¢ð¥ð╣ ðÿðíðó ð┐ð░Ðåð©ðÁð¢Ðéð║ðÁ ð▒Ðïð╗ð░ ð▓Ðïð┐ð¥ð╗ð¢ðÁð¢ð░ ð┤ðÁð¢Ðüð©Ðéð¥ð╝ðÁÐéÐÇð©ÐÅ, ð┐ð¥┬áÐÇðÁðÀÐâð╗ÐîÐéð░Ðéð░ð╝ ð║ð¥Ðéð¥ÐÇð¥ð╣ ð▓ÐïÐÅð▓ð╗ðÁð¢ð░ ð¥ÐüÐéðÁð¥ð┐ðÁð¢ð©ÐÅ ÐêðÁð╣ð║ð© ð▒ðÁð┤ÐÇðÁð¢ð¢ð¥ð╣ ð║ð¥ÐüÐéð© ð©┬áð¥ÐüÐéðÁð¥ð┐ð¥ÐÇð¥ðÀ ð┐ð¥ÐÅÐüð¢ð©Ðçð¢ð¥ð│ð¥ ð¥Ðéð┤ðÁð╗ð░ ð┐ð¥ðÀð▓ð¥ð¢ð¥Ðçð¢ð©ð║ð░; Ðìð¢ð┤ð¥ð║ÐÇð©ð¢ð¥ð╗ð¥ð│ð¥ð╝ ð¢ð░ðÀð¢ð░ÐçðÁð¢ð¥ ð╗ðÁÐçðÁð¢ð©ðÁ ð┐ÐÇðÁð┐ð░ÐÇð░Ðéð░ð╝ð© ð░ð╗ðÁð¢ð┤ÐÇð¥ð¢ð¥ð▓ð¥ð╣ ð║ð©Ðüð╗ð¥ÐéÐï ð©┬áð▓ð©Ðéð░ð╝ð©ð¢ð¥ð╝ D Ðü┬áð┐ð¥ð╗ð¥ðÂð©ÐéðÁð╗Ðîð¢Ðïð╝ ÐìÐäÐäðÁð║Ðéð¥ð╝.
ðÆ┬áð║ð¥ð░ð│Ðâð╗ð¥ð│ÐÇð░ð╝ð╝ðÁ, ð║ð╗ð©ð¢ð©ÐçðÁÐüð║ð¥ð╝ ð©┬áð▒ð©ð¥Ðàð©ð╝ð©ÐçðÁÐüð║ð¥ð╝ ð░ð¢ð░ð╗ð©ðÀð░Ðà ð║ÐÇð¥ð▓ð© ÔÇô ð▒ðÁðÀ ð¥Ðéð║ð╗ð¥ð¢ðÁð¢ð©ð╣; Ðüð¥ÐàÐÇð░ð¢ÐÅð╗ÐüÐÅ ð¢ð¥ÐÇð╝ð░ð╗Ðîð¢Ðïð╣ ÐâÐÇð¥ð▓ðÁð¢Ðî IgG (Ðéð░ð▒ð╗.┬á1).
ðöð╗ÐÅ ÐÇðÁÐêðÁð¢ð©ÐÅ ð▓ð¥ð┐ÐÇð¥Ðüð░ ð¥ð▒┬áð¥Ðéð╝ðÁð¢ðÁ ðÿðíðó ÐéÐÇðÁð▒ð¥ð▓ð░ð╗ð¥ÐüÐî ð┐ÐÇð¥ð▓ðÁð┤ðÁð¢ð©ðÁ ð║ð¥ð¢ÐéÐÇð¥ð╗Ðîð¢ð¥ð╣ ð▒ð©ð¥ð┐Ðüð©ð© ð┐ðÁÐçðÁð¢ð©, Ðéð░ð║ ð║ð░ð║ ð¥ÐéÐüÐâÐéÐüÐéð▓ð©ðÁ ð░ð║Ðéð©ð▓ð¢ð¥ÐüÐéð© ð╗ð░ð▒ð¥ÐÇð░Ðéð¥ÐÇð¢ð¥ ð¢ðÁ┬áð┐ð¥ðÀð▓ð¥ð╗ÐÅðÁÐé ð┤ð¥ÐüÐéð¥ð▓ðÁÐÇð¢ð¥ ÐüÐâð┤ð©ÐéÐî ð¥┬áð¢ð░ð╗ð©Ðçð©ð© ð│ð©ÐüÐéð¥ð╗ð¥ð│ð©ÐçðÁÐüð║ð¥ð╣ ÐÇðÁð╝ð©ÐüÐüð©ð©. ðƒÐÇð¥Ðéð©ð▓ð¥ð┐ð¥ð║ð░ðÀð░ð¢ð©ð╣ ð║┬áð┐ÐÇð¥ÐåðÁð┤ÐâÐÇðÁ ð¢ðÁ┬áð▒Ðïð╗ð¥, ð┐ð░Ðåð©ðÁð¢Ðéð║ð░ ð┤ð░ð╗ð░ ð©ð¢Ðäð¥ÐÇð╝ð©ÐÇð¥ð▓ð░ð¢ð¢ð¥ðÁ Ðüð¥ð│ð╗ð░Ðüð©ðÁ; ð┐ÐÇð¥ð▓ðÁð┤ðÁð¢ð░ Ðéð¥ð¢ð║ð¥ð©ð│ð¥ð╗Ðîð¢ð░ÐÅ ð┐Ðâð¢ð║Ðåð©ð¥ð¢ð¢ð░ÐÅ ð▒ð©ð¥ð┐Ðüð©ÐÅ ð┐ðÁÐçðÁð¢ð©, ð┐ð¥ð╗ÐâÐçðÁð¢ ÐüÐéð¥ð╗ð▒ð©ð║ Ðéð║ð░ð¢ð© ð┤ð╗ð©ð¢ð¥ð╣ ┬á18 ð╝ð╝. ðƒð¥┬áÐÇðÁðÀÐâð╗ÐîÐéð░Ðéð░ð╝ ð©ÐüÐüð╗ðÁð┤ð¥ð▓ð░ð¢ð©ÐÅ ð▒ð©ð¥ð┐Ðéð░Ðéð░ ð┐ðÁÐçðÁð¢ð© ð▒Ðïð╗ð¥ ð┐ð¥ð╗ÐâÐçðÁð¢ð¥ ðÀð░ð║ð╗ÐÄÐçðÁð¢ð©ðÁ ð╝ð¥ÐÇÐäð¥ð╗ð¥ð│ð░: ┬½ðÉÐâÐéð¥ð©ð╝ð╝Ðâð¢ð¢Ðïð╣ ð│ðÁð┐ð░Ðéð©Ðé ð¢ð©ðÀð║ð¥ð╣ ÐüÐéðÁð┐ðÁð¢ð© ð░ð║Ðéð©ð▓ð¢ð¥ÐüÐéð©. ðƒðÁÐÇð▓ð©Ðçð¢Ðïð╣ ð▒ð©ð╗ð©ð░ÐÇð¢Ðïð╣ Ðàð¥ð╗ð░ð¢ð│ð©Ðé. ðƒÐÇð© ÐüÐÇð░ð▓ð¢ðÁð¢ð©ð© Ðü┬áð┐ÐÇðÁð┐ð░ÐÇð░Ðéð░ð╝ð© ð¥Ðé┬á2021 ð│.┬á ð¢ð░ð▒ð╗ÐÄð┤ð░ðÁÐéÐüÐÅ ð┐ð¥ð╗ð¥ðÂð©ÐéðÁð╗Ðîð¢ð░ÐÅ ð┤ð©ð¢ð░ð╝ð©ð║ð░, ð║ð¥Ðéð¥ÐÇð░ÐÅ ðÀð░ð║ð╗ÐÄÐçð░ðÁÐéÐüÐÅ ð▓┬áÐüð¢ð©ðÂðÁð¢ð©ð© ð░ð║Ðéð©ð▓ð¢ð¥ÐüÐéð© ð┐ð¥ÐÇÐéð░ð╗Ðîð¢ð¥ð│ð¥ ð▓ð¥Ðüð┐ð░ð╗ðÁð¢ð©ÐÅ, ð¥ÐéÐüÐâÐéÐüÐéð▓ð©ð© ÐüÐéÐâð┐ðÁð¢Ðçð░ÐéÐïÐà ð¢ðÁð║ÐÇð¥ðÀð¥ð▓. ðÿðôðÉ ð┐ð¥┬áKnodell ÔÇô 4┬╗.
ðƒð¥ð╗ð¢ð░ÐÅ ð¥Ðéð╝ðÁð¢ð░ ðÿðíðó ð┐ÐÇð© ðÉðÿðô ð┐ð¥ð║ð░ðÀð░ð¢ð░ ð┐ÐÇð© Ðüð¢ð©ðÂðÁð¢ð©ð© ðÿðôðÉ ð┤ð¥ 3 ð▒ð░ð╗ð╗ð¥ð▓ ð©┬áð╝ðÁð¢ðÁðÁ. ðÆ┬áð┤ð░ð¢ð¢ð¥ð╝ ð║ð╗ð©ð¢ð©ÐçðÁÐüð║ð¥ð╝ Ðüð╗ÐâÐçð░ðÁ Ðü┬áÐâÐçðÁÐéð¥ð╝ ð┤ð¥ÐüÐéð░Ðéð¥Ðçð¢ð¥ð│ð¥ ð┐ðÁÐÇð©ð¥ð┤ð░ ÐéðÁÐÇð░ð┐ð©ð© ð┐ð¥ð│ÐÇð░ð¢ð©Ðçð¢ÐïÐà ð┐ð¥ð║ð░ðÀð░ÐéðÁð╗ðÁð╣ ðÿðôðÉ ð©┬áÐÇð░ðÀð▓ð©Ðéð©ÐÅ ð¢ðÁðÂðÁð╗ð░ÐéðÁð╗Ðîð¢ÐïÐà ÐÅð▓ð╗ðÁð¢ð©ð╣ ð▓┬áð▓ð©ð┤ðÁ ð¥ÐüÐéðÁð¥ð┐ð¥ÐÇð¥ðÀð░ ð▒Ðïð╗ð░ ð©ðÀð▒ÐÇð░ð¢ð░ Ðéð░ð║Ðéð©ð║ð░ ÐüÐéÐâð┐ðÁð¢Ðçð░Ðéð¥ð╣ ð¥Ðéð╝ðÁð¢Ðï ð©ð╝ð╝Ðâð¢ð¥ÐüÐâð┐ÐÇðÁÐüÐüð©ð©: ð╝ðÁÐéð©ð╗ð┐ÐÇðÁð┤ð¢ð©ðÀð¥ð╗ð¥ð¢ ð▒Ðïð╗ ð┐ð¥ÐüÐéðÁð┐ðÁð¢ð¢ð¥ ð¥Ðéð╝ðÁð¢ðÁð¢, ð▓┬áÐéð¥ ð▓ÐÇðÁð╝ÐÅ ð║ð░ð║ ð┐ÐÇð©ðÁð╝ ð░ðÀð░Ðéð©ð¥ð┐ÐÇð©ð¢ð░ ð▒Ðïð╗ ð┐ÐÇð¥ð┤ð¥ð╗ðÂðÁð¢ ð¢ð░┬áÐüÐÇð¥ð║ ÐêðÁÐüÐéÐî ð╝ðÁÐüÐÅÐåðÁð▓ Ðü┬áð┐ð¥Ðüð╗ðÁð┤ÐâÐÄÐëðÁð╣ ð┐ð¥ð╗ð¢ð¥ð╣ ðÁð│ð¥ ð¥Ðéð╝ðÁð¢ð¥ð╣.
ðæð©ð¥Ðàð©ð╝ð©ÐçðÁÐüð║ð░ÐÅ ÐÇðÁð╝ð©ÐüÐüð©ÐÅ Ðüð¥ÐàÐÇð░ð¢ÐÅðÁÐéÐüÐÅ ð¢ð░┬áð▓ÐüðÁÐà ÐìÐéð░ð┐ð░Ðà ð¢ð░ð▒ð╗ÐÄð┤ðÁð¢ð©ÐÅ: ÐçðÁÐÇðÁðÀ ÐéÐÇð© ð╝ðÁÐüÐÅÐåð░ ð┐ð¥Ðüð╗ðÁ ð¥Ðéð╝ðÁð¢Ðï ð╝ðÁÐéð©ð╗ð┐ÐÇðÁð┤ð¢ð©ðÀð¥ð╗ð¥ð¢ð░ ð©┬áÐçðÁÐÇðÁðÀ ð▓ð¥ÐüðÁð╝Ðî ð╝ðÁÐüÐÅÐåðÁð▓ ð┐ð¥Ðüð╗ðÁ ð¥Ðéð╝ðÁð¢Ðï ð░ðÀð░Ðéð©ð¥ð┐ÐÇð©ð¢ð░.
ð×ð▒ÐüÐâðÂð┤ðÁð¢ð©ðÁ
ðØð░ÐüÐéð¥ÐÅÐëðÁðÁ ð║ð╗ð©ð¢ð©ÐçðÁÐüð║ð¥ðÁ ð¢ð░ð▒ð╗ÐÄð┤ðÁð¢ð©ðÁ ð┐ð¥ðÀð▓ð¥ð╗ÐÅðÁÐé ð▓Ðïð┤ðÁð╗ð©ÐéÐî ð¢ðÁÐüð║ð¥ð╗Ðîð║ð¥ ð║ð╗ÐÄÐçðÁð▓ÐïÐà ð░Ðüð┐ðÁð║Ðéð¥ð▓ ð▓ðÁð┤ðÁð¢ð©ÐÅ ð┐ð░Ðåð©ðÁð¢Ðéð¥ð▓ Ðü┬áðÉðÿðùðƒ:
- ð▓ð░ðÂð¢ð¥ÐüÐéÐî ÐÇð░ÐüÐêð©ÐÇðÁð¢ð¢ð¥ð│ð¥ ð┤ð©ð░ð│ð¢ð¥ÐüÐéð©ÐçðÁÐüð║ð¥ð│ð¥ ð┐ð¥ð©Ðüð║ð░┬áð┐ÐÇð© ð┐ðÁÐÇð▓ð©Ðçð¢ð¥ð╝ ð▓ÐïÐÅð▓ð╗ðÁð¢ð©ð© Ðåð©Ðéð¥ð╗ð©ðÀð░ ð©/ð©ð╗ð© Ðàð¥ð╗ðÁÐüÐéð░ðÀð░ ð┤ð╗ÐÅ Ðüð▓ð¥ðÁð▓ÐÇðÁð╝ðÁð¢ð¢ð¥ð│ð¥ ð▓ÐïÐÅð▓ð╗ðÁð¢ð©ÐÅ ÐìÐéð©ð¥ð╗ð¥ð│ð©ð© ðÀð░ð▒ð¥ð╗ðÁð▓ð░ð¢ð©ÐÅ;
- ÐÇð¥ð╗Ðî ð╝ð¥ÐÇÐäð¥ð╗ð¥ð│ð©ÐçðÁÐüð║ð¥ð│ð¥ ð©ÐüÐüð╗ðÁð┤ð¥ð▓ð░ð¢ð©ÐÅ ð┐ðÁÐçðÁð¢ð©;
- ð┤ð©ð░ð│ð¢ð¥ÐüÐéð©ÐçðÁÐüð║ð░ÐÅ Ðéð¥Ðçð¢ð¥ÐüÐéÐî Ðäð©ð▒ÐÇð¥Ðìð╗ð░ÐüÐéð¥ð╝ðÁÐéÐÇð©ð© ð┐ÐÇð© ðÉðÿðùðƒ;
- ð║ÐÇð©ÐéðÁÐÇð©ð© ð¥Ðéð╝ðÁð¢Ðï ðÿðíðó.
ðÆ┬áð┐ÐÇðÁð┤ÐüÐéð░ð▓ð╗ðÁð¢ð¢ð¥ð╝ ð║ð╗ð©ð¢ð©ÐçðÁÐüð║ð¥ð╝ ð¢ð░ð▒ð╗ÐÄð┤ðÁð¢ð©ð© ð▒Ðïð╗ ð┤ð©ð░ð│ð¢ð¥ÐüÐéð©ÐÇð¥ð▓ð░ð¢ ð▓ð░ÐÇð©ð░ð¢Ðéð¢Ðïð╣ Ðüð©ð¢ð┤ÐÇð¥ð╝ ┬½ðƒðæðÑ, ðÉð£ðÉ-ð┐ð¥ðÀð©Ðéð©ð▓ð¢Ðïð╣, Ðü┬áð┐ÐÇð©ðÀð¢ð░ð║ð░ð╝ð© ðÉðÿðô┬╗, ð¢ð░ðÀð¢ð░ÐçðÁð¢ð░ ð░ð┤ðÁð║ð▓ð░Ðéð¢ð░ÐÅ ÐéðÁÐÇð░ð┐ð©ÐÅ, ð┤ð¥ÐüÐéð©ð│ð¢ÐâÐéð░ ÐüÐéð¥ð╣ð║ð░ÐÅ ÐÇðÁð╝ð©ÐüÐüð©ÐÅ ðÀð░ð▒ð¥ð╗ðÁð▓ð░ð¢ð©ÐÅ. ð×ð┤ð¢ð░ð║ð¥ ð¥Ðé┬áð╝ð¥ð╝ðÁð¢Ðéð░ ð┐ðÁÐÇð▓ð©Ðçð¢ð¥ð│ð¥ ð▓ÐïÐÅð▓ð╗ðÁð¢ð©ÐÅ ð©ðÀð╝ðÁð¢ðÁð¢ð©ð╣ ð▓┬áð▒ð©ð¥Ðàð©ð╝ð©ÐçðÁÐüð║ð©Ðà ð┐ð¥ð║ð░ðÀð░ÐéðÁð╗ÐÅÐà ð║ÐÇð¥ð▓ð© ð┤ð¥ ÐâÐüÐéð░ð¢ð¥ð▓ð╗ðÁð¢ð©ÐÅ ð¥ð║ð¥ð¢Ðçð░ÐéðÁð╗Ðîð¢ð¥ð│ð¥ ð║ð╗ð©ð¢ð©ÐçðÁÐüð║ð¥ð│ð¥ ð┤ð©ð░ð│ð¢ð¥ðÀð░ Ðâ┬áð┤ð░ð¢ð¢ð¥ð╣ ð┐ð░Ðåð©ðÁð¢Ðéð║ð© ð┐ÐÇð¥Ðêð╗ð¥ ð▒ð¥ð╗ðÁðÁ ð┤ð▓ÐâÐà ð╗ðÁÐé.
ð¡Ðéð¥Ðé ð┐ÐÇð©ð╝ðÁÐÇ ð¢ð░ð│ð╗ÐÅð┤ð¢ð¥ ð┤ðÁð╝ð¥ð¢ÐüÐéÐÇð©ÐÇÐâðÁÐé Ðàð░ÐÇð░ð║ÐéðÁÐÇð¢ÐâÐÄ ð┤ð╗ÐÅ ð┤ð░ð¢ð¢ð¥ð╣ ð┐ð░Ðéð¥ð╗ð¥ð│ð©ð© ð┐ÐÇð¥ð▒ð╗ðÁð╝Ðâ ð┐ð¥ðÀð┤ð¢ðÁð╣ ð┤ð©ð░ð│ð¢ð¥ÐüÐéð©ð║ð©, ÐçÐéð¥ ð┐ð¥ð┤Ðéð▓ðÁÐÇðÂð┤ð░ðÁÐéÐüÐÅ ð¢ð░Ðêð©ð╝ Ðüð¥ð▒ÐüÐéð▓ðÁð¢ð¢Ðïð╝ ð¥ð┐ÐïÐéð¥ð╝ ð▓ðÁð┤ðÁð¢ð©ÐÅ ð┐ð░Ðåð©ðÁð¢Ðéð¥ð▓ Ðü┬áðÉðÿðùðƒ ð©┬áÐüð¥ð│ð╗ð░ÐüÐâðÁÐéÐüÐÅ Ðü┬áð┤ð░ð¢ð¢Ðïð╝ð© ð╗ð©ÐéðÁÐÇð░ÐéÐâÐÇÐï.
ðÿð╗ð╗ÐÄÐüÐéÐÇð░Ðåð©ðÁð╣ ð╝ð¥ðÂðÁÐé Ðüð╗ÐâðÂð©ÐéÐî ð©ÐüÐüð╗ðÁð┤ð¥ð▓ð░ð¢ð©ðÁ, ð¥ð┐Ðâð▒ð╗ð©ð║ð¥ð▓ð░ð¢ð¢ð¥ðÁ ð▓┬áðÂÐâÐÇð¢ð░ð╗ðÁ JHEP Reports ð▓┬á2024 ð│., ð▓┬áð║ð¥Ðéð¥ÐÇð¥ð╝ ð┐ÐÇð¥ð░ð¢ð░ð╗ð©ðÀð©ÐÇð¥ð▓ð░ð¢Ðï ð┤ð░ð¢ð¢ÐïðÁ 1331 ð┐ð░Ðåð©ðÁð¢Ðéð░ Ðü┬áðÉðÿðùðƒ, ð©ðÀ┬áð¢ð©Ðà ┬áÐâ┬á83 ÐçðÁð╗ð¥ð▓ðÁð║ ÐâÐüÐéð░ð¢ð¥ð▓ð╗ðÁð¢ ð▓ð░ÐÇð©ð░ð¢Ðéð¢Ðïð╣ Ðüð©ð¢ð┤ÐÇð¥ð╝ ðƒðæðÑ Ðü┬áð┐ÐÇð©ðÀð¢ð░ð║ð░ð╝ð© ðÉðÿðô. ðíð©ð¢ÐàÐÇð¥ð¢ð¢ð░ÐÅ ð┤ð©ð░ð│ð¢ð¥ÐüÐéð©ð║ð░ ðƒðæðÑ ð©┬áðÉðÿðô ð▒Ðïð╗ð░ ðÀð░Ðäð©ð║Ðüð©ÐÇð¥ð▓ð░ð¢ð░ ð╗ð©ÐêÐî Ðâ┬á56 (67%) ð▒ð¥ð╗Ðîð¢ÐïÐà. ðú┬áð¥ÐüÐéð░ð╗Ðîð¢ÐïÐà 27 (33%) ð┐ð░Ðåð©ðÁð¢Ðéð¥ð▓ ð┤ð©ð░ð│ð¢ð¥ðÀ ÐâÐüÐéð░ð¢ð░ð▓ð╗ð©ð▓ð░ð╗ÐüÐÅ ð┐ð¥ÐìÐéð░ð┐ð¢ð¥: ð©ðÀ┬áð¢ð©Ðà Ðâ┬á22 ð©ðÀð¢ð░Ðçð░ð╗Ðîð¢ð¥ ð▒Ðïð╗ ð▓ðÁÐÇð©Ðäð©Ðåð©ÐÇð¥ð▓ð░ð¢ ðƒðæðÑ, ð░┬áÐâ 5 ÔÇô ┬áðÉðÿðô. ð×ð▒ÐÇð░Ðëð░ðÁÐé ð¢ð░┬áÐüðÁð▒ÐÅ ð▓ð¢ð©ð╝ð░ð¢ð©ðÁ ð┤ð╗ð©ÐéðÁð╗Ðîð¢Ðïð╣ ð╝ðÁð┤ð©ð░ð¢ð¢Ðïð╣ ð©ð¢ÐéðÁÐÇð▓ð░ð╗ ð╝ðÁðÂð┤Ðâ ð┐ðÁÐÇð▓Ðïð╝ ð┐ÐÇð¥ÐÅð▓ð╗ðÁð¢ð©ðÁð╝ ð▒ð¥ð╗ðÁðÀð¢ð© ð©┬áð┐ð¥ÐüÐéð░ð¢ð¥ð▓ð║ð¥ð╣ ð¥ð║ð¥ð¢Ðçð░ÐéðÁð╗Ðîð¢ð¥ð│ð¥ ð┤ð©ð░ð│ð¢ð¥ðÀð░, Ðüð¥ÐüÐéð░ð▓ð©ð▓Ðêð©ð╣ ÐçðÁÐéÐïÐÇðÁ ð│ð¥ð┤ð░ (ð┤ð©ð░ð┐ð░ðÀð¥ð¢ ð¥Ðé┬áÐüðÁð╝ð© ð╝ðÁÐüÐÅÐåðÁð▓ ð┤ð¥ 16 ð╗ðÁÐé) [8]. ð¡Ðéð¥ ð┐ð¥ð┤ÐçðÁÐÇð║ð©ð▓ð░ðÁÐé ð║ð░ð║ Ðüð╗ð¥ðÂð¢ð¥ÐüÐéÐî ð┤ð©ð░ð│ð¢ð¥ÐüÐéð©ð║ð© ð©┬áÐçð░ÐüÐéð¥ ð┐ð¥ðÀð┤ð¢ðÁðÁ ÐÇð░Ðüð┐ð¥ðÀð¢ð░ð▓ð░ð¢ð©ðÁ ð┤ð░ð¢ð¢ð¥ð╣ Ðäð¥ÐÇð╝Ðï ð┐ð░Ðéð¥ð╗ð¥ð│ð©ð©, Ðéð░ð║ ð©┬áðÁðÁ ð▓ð¥ðÀð╝ð¥ðÂð¢Ðïð╣ ð┐ð¥Ðüð╗ðÁð┤ð¥ð▓ð░ÐéðÁð╗Ðîð¢Ðïð╣ ð▓ð░ÐÇð©ð░ð¢Ðé ÐÇð░ðÀð▓ð©Ðéð©ÐÅ, ð┐ÐÇð© ð║ð¥Ðéð¥ÐÇð¥ð╝ ðÀð░ð▒ð¥ð╗ðÁð▓ð░ð¢ð©ðÁ ð╝ð¥ðÂðÁÐé ð┤ðÁð▒ÐÄÐéð©ÐÇð¥ð▓ð░ÐéÐî ð║ð░ð║ Ðü┬áðƒðæðÑ, Ðéð░ð║ ð©┬áÐü ðÉðÿðô, Ðü┬áð┐ð¥Ðüð╗ðÁð┤ÐâÐÄÐëð©ð╝ ð┐ÐÇð©Ðüð¥ðÁð┤ð©ð¢ðÁð¢ð©ðÁð╝ ð┐ÐÇð©ðÀð¢ð░ð║ð¥ð▓ ð▓Ðéð¥ÐÇð¥ð╣ ð¢ð¥ðÀð¥ð╗ð¥ð│ð©ð©.
ðÆ┬áð┐ÐÇðÁð┤ÐüÐéð░ð▓ð╗ðÁð¢ð¢ð¥ð╝ ð║ð╗ð©ð¢ð©ÐçðÁÐüð║ð¥ð╝ ð¢ð░ð▒ð╗ÐÄð┤ðÁð¢ð©ð© ð▓┬áÐéðÁÐçðÁð¢ð©ðÁ ð┐ð¥ð╗ÐâÐéð¥ÐÇð░ ð╗ðÁÐé ð╗ðÁÐçðÁð¢ð©ðÁ ð┐ð░Ðåð©ðÁð¢Ðéð║ð© ð▓┬áð░ð╝ð▒Ðâð╗ð░Ðéð¥ÐÇð¢ÐïÐà ÐâÐüð╗ð¥ð▓ð©ÐÅÐà ð¥ÐüÐâÐëðÁÐüÐéð▓ð╗ÐÅð╗ð¥ÐüÐî Ðü┬áÐäð¥ÐÇð╝Ðâð╗ð©ÐÇð¥ð▓ð║ð¥ð╣ ┬½ÐàÐÇð¥ð¢ð©ÐçðÁÐüð║ð©ð╣ ð│ðÁð┐ð░Ðéð©Ðé ð¢ðÁÐâÐéð¥Ðçð¢ðÁð¢ð¢ð¥ð╣ ÐìÐéð©ð¥ð╗ð¥ð│ð©ð©┬╗ ð▒ðÁðÀ ð┐ÐÇð¥ð▓ðÁð┤ðÁð¢ð©ÐÅ Ðâð│ð╗Ðâð▒ð╗ðÁð¢ð¢ð¥ð│ð¥ ð¥ð▒Ðüð╗ðÁð┤ð¥ð▓ð░ð¢ð©ÐÅ ð┤ð╗ÐÅ ÐâÐüÐéð░ð¢ð¥ð▓ð╗ðÁð¢ð©ÐÅ ð│ðÁð¢ðÁðÀð░ ðÀð░ð▒ð¥ð╗ðÁð▓ð░ð¢ð©ÐÅ.
ð×ð┐ð©Ðüð░ð¢ð¢ð░ÐÅ Ðüð©ÐéÐâð░Ðåð©ÐÅ ð▓ÐïÐüð▓ðÁÐçð©ð▓ð░ðÁÐé ð©┬áð┤ÐÇÐâð│ÐâÐÄ ð║ð╗ÐÄÐçðÁð▓ÐâÐÄ ð║ð╗ð©ð¢ð©ÐçðÁÐüð║ÐâÐÄ ð┐ÐÇð¥ð▒ð╗ðÁð╝Ðâ: ð┐ÐÇð© ð¥ð▒ð¢ð░ÐÇÐâðÂðÁð¢ð©ð© ð¢ðÁð¥ð▒ÐèÐÅÐüð¢ð©ð╝ð¥ð│ð¥ Ðåð©Ðéð¥ð╗ð©ðÀð░ ÐéÐÇðÁð▒ÐâðÁÐéÐüÐÅ ð┐ÐÇð¥ð▓ðÁð┤ðÁð¢ð©ðÁ ÐÇð░ÐüÐêð©ÐÇðÁð¢ð¢ð¥ð│ð¥ ð┤ð©ð░ð│ð¢ð¥ÐüÐéð©ÐçðÁÐüð║ð¥ð│ð¥ ð┐ð¥ð©Ðüð║ð░ ð┤ð╗ÐÅ ÐâÐüÐéð░ð¢ð¥ð▓ð╗ðÁð¢ð©ÐÅ ÐìÐéð©ð¥ð╗ð¥ð│ð©ÐçðÁÐüð║ð¥ð│ð¥ Ðäð░ð║Ðéð¥ÐÇð░. ðÆ┬áð┤ð©ÐäÐäðÁÐÇðÁð¢Ðåð©ð░ð╗Ðîð¢Ðïð╣ ÐÇÐÅð┤ ð┤ð¥ð╗ðÂð¢Ðï ð▒ÐïÐéÐî ð▓ð║ð╗ÐÄÐçðÁð¢Ðï ð░ÐâÐéð¥ð©ð╝ð╝Ðâð¢ð¢ÐïðÁ ð┐ÐÇð¥ÐåðÁÐüÐüÐï, ð▒ð¥ð╗ðÁðÀð¢ð© ð¢ð░ð║ð¥ð┐ð╗ðÁð¢ð©ÐÅ, ð▓ð¥ðÀð┤ðÁð╣ÐüÐéð▓ð©ðÁ ð│ðÁð┐ð░Ðéð¥Ðéð¥ð║Ðüð©Ðçð¢ÐïÐà ð░ð│ðÁð¢Ðéð¥ð▓ ð©┬áð┤ÐÇÐâð│ð©ðÁ ð┐ÐÇð©Ðçð©ð¢Ðï, ð░┬áð¢ðÁ Ðéð¥ð╗Ðîð║ð¥ ð©Ðüð║ð╗ÐÄÐçðÁð¢ð©ðÁ ð▓ð©ÐÇÐâÐüð¢ÐïÐà ð│ðÁð┐ð░Ðéð©Ðéð¥ð▓, ð║ð░ð║ ÐìÐéð¥ ð©ð╝ðÁð╗ð¥ ð╝ðÁÐüÐéð¥ ð▓┬áð¥ð┐ð©Ðüð░ð¢ð¢ð¥ð╝ Ðüð╗ÐâÐçð░ðÁ.
ðÆð╝ðÁÐüÐéðÁ Ðü┬áÐéðÁð╝ ÐâðÂðÁ ð┐ÐÇð© ð┐ðÁÐÇð▓ð¥ð╝ ð¥ð▒Ðüð╗ðÁð┤ð¥ð▓ð░ð¢ð©ð© ð▓ÐïÐÅð▓ð╗ÐÅð╗ÐüÐÅ Ðüð©ð¢ð┤ÐÇð¥ð╝ Ðàð¥ð╗ðÁÐüÐéð░ðÀð░ (ð║ð░ð║ ð╗ð░ð▒ð¥ÐÇð░Ðéð¥ÐÇð¢ð¥ ÔÇô ð┐ð¥ð▓ÐïÐêðÁð¢ð©ðÁ ÐâÐÇð¥ð▓ð¢ÐÅ ð®ðñ ð┤ð¥ 3 ðÆðôðØ, Ðéð░ð║ ð©┬áð║ð╗ð©ð¢ð©ÐçðÁÐüð║ð© ÔÇô ð▓┬áð▓ð©ð┤ðÁ ð║ð¥ðÂð¢ð¥ð│ð¥ ðÀÐâð┤ð░), ð▓┬áÐüð▓ÐÅðÀð© Ðü┬áÐçðÁð╝ ÐéÐÇðÁð▒ð¥ð▓ð░ð╗ð¥ÐüÐî ð©Ðüð║ð╗ÐÄÐçðÁð¢ð©ðÁ Ðàð¥ð╗ðÁÐüÐéð░Ðéð©ÐçðÁÐüð║ð©Ðà ðÀð░ð▒ð¥ð╗ðÁð▓ð░ð¢ð©ð╣ ð┐ðÁÐçðÁð¢ð©, ð¢ð░ð©ð▒ð¥ð╗ðÁðÁ Ðçð░ÐüÐéÐïð╝ð© ð©ðÀ┬áð║ð¥Ðéð¥ÐÇÐïÐà ÐÅð▓ð╗ÐÅÐÄÐéÐüÐÅ ðƒðæðÑ ð©┬áðƒðíðÑ. ðƒð¥ÐìÐéð¥ð╝Ðâ ð▓ÐüðÁð╝ ð┐ð░Ðåð©ðÁð¢Ðéð░ð╝ Ðü┬áÐüð©ð¢ð┤ÐÇð¥ð╝ð¥ð╝ Ðàð¥ð╗ðÁÐüÐéð░ðÀð░ ÐÇðÁð║ð¥ð╝ðÁð¢ð┤ÐâðÁÐéÐüÐÅ ð¥ÐåðÁð¢ð©ð▓ð░ÐéÐî Ðéð©ÐéÐÇÐï ð░ÐâÐéð¥ð░ð¢Ðéð©ÐéðÁð╗ ðÉð£ðÉ-ð£2, sp-100 ð©┬ágp-210 ð©┬áð▓Ðïð┐ð¥ð╗ð¢ÐÅÐéÐî ð╝ð░ð│ð¢ð©Ðéð¢ð¥-ÐÇðÁðÀð¥ð¢ð░ð¢Ðüð¢ÐâÐÄ Ðàð¥ð╗ð░ð¢ð│ð©ð¥ð┐ð░ð¢ð║ÐÇðÁð░Ðéð¥ð│ÐÇð░Ðäð©ÐÄ (ð£ðáðÑðƒðô) [9].
ðöÐÇÐâð│ð©ð╝ð© ð▓ð¥ðÀð╝ð¥ðÂð¢Ðïð╝ð© ð┐ÐÇð©Ðçð©ð¢ð░ð╝ð© Ðàð¥ð╗ðÁÐüÐéð░Ðéð©ÐçðÁÐüð║ð¥ð│ð¥ Ðüð©ð¢ð┤ÐÇð¥ð╝ð░ ð╝ð¥ð│ÐâÐé ð▒ÐïÐéÐî ð╗ðÁð║ð░ÐÇÐüÐéð▓ðÁð¢ð¢ÐïðÁ ð┐ð¥ÐÇð░ðÂðÁð¢ð©ÐÅ ð┐ðÁÐçðÁð¢ð©, IgG4-ð░ÐüÐüð¥Ðåð©ð©ÐÇð¥ð▓ð░ð¢ð¢Ðïð╣ Ðàð¥ð╗ð░ð¢ð│ð©Ðé, ð░┬áÐéð░ð║ðÂðÁ ð▓ð©ÐÇÐâÐüð¢Ðïð╣ ð│ðÁð┐ð░Ðéð©Ðé: ð▓ð░ðÂð¢ð¥ ð┐ð¥ð╝ð¢ð©ÐéÐî ð¥┬áð▓ð¥ðÀð╝ð¥ðÂð¢ð¥ð╝ ð©ð¢Ðäð©Ðåð©ÐÇð¥ð▓ð░ð¢ð©ð© ð▓ð©ÐÇÐâÐüð░ð╝ð© ð│ðÁð┐ð░Ðéð©Ðéð¥ð▓ ðÉ┬áð© ðò, ð░┬áÐéð░ð║ðÂðÁ ð¥┬áÐÇðÁð┤ð║ð©Ðà Ðüð╗ÐâÐçð░ÐÅÐà ð┐ð¥ÐÇð░ðÂðÁð¢ð©ÐÅ ð┐ðÁÐçðÁð¢ð© ð▓ð©ÐÇÐâÐüð░ð╝ð©, ÐéÐÇð░ð┤ð©Ðåð©ð¥ð¢ð¢ð¥ ð¢ðÁ┬áð¥Ðéð¢ð¥ÐüÐÅÐëð©ð╝ð©ÐüÐÅ ð║┬áð│ðÁð┐ð░Ðéð¥ÐéÐÇð¥ð┐ð¢Ðïð╝ (ð¢ð░ð┐ÐÇð©ð╝ðÁÐÇ, Ðåð©Ðéð¥ð╝ðÁð│ð░ð╗ð¥ð▓ð©ÐÇÐâÐü ð©┬áð┤ÐÇ.). ð×ÐéÐüÐâÐéÐüÐéð▓ð©ðÁ ð¢ðÁð¥ð▒Ðàð¥ð┤ð©ð╝ÐïÐà ð┤ð©ð░ð│ð¢ð¥ÐüÐéð©ÐçðÁÐüð║ð©Ðà ð╝ðÁÐÇð¥ð┐ÐÇð©ÐÅÐéð©ð╣ ð┐ÐÇð©ð▓ð¥ð┤ð©Ðé ð║┬áðÀð░ð┐ð¥ðÀð┤ð░ð╗ð¥ð╣ ð┤ð©ð░ð│ð¢ð¥ÐüÐéð©ð║ðÁ, ð¢ðÁÐìÐäÐäðÁð║Ðéð©ð▓ð¢ð¥ð╝Ðâ ð╗ðÁÐçðÁð¢ð©ÐÄ ð│ðÁð┐ð░Ðéð¥ð┐ÐÇð¥ÐéðÁð║Ðéð¥ÐÇð¢Ðïð╝ð© ð┐ÐÇðÁð┐ð░ÐÇð░Ðéð░ð╝ð©, ð╝ð¢ð¥ð│ð©ðÁ ð©ðÀ┬áð║ð¥Ðéð¥ÐÇÐïÐà ð¢ðÁ┬áð©ð╝ðÁÐÄÐé ð┤ð¥ÐüÐéð░Ðéð¥Ðçð¢ð¥ð╣ ð┤ð¥ð║ð░ðÀð░ÐéðÁð╗Ðîð¢ð¥ð╣ ð▒ð░ðÀÐï, ð©┬áð┐ÐÇð¥ð│ÐÇðÁÐüÐüð©ÐÇð¥ð▓ð░ð¢ð©ÐÄ Ðäð©ð▒ÐÇð¥ðÀð░ ð┐ðÁÐçðÁð¢ð©.
ðÆ┬áð┤ð░ð¢ð¢ð¥ð╝ ð║ð╗ð©ð¢ð©ÐçðÁÐüð║ð¥ð╝ ð┐ÐÇð©ð╝ðÁÐÇðÁ Ðéð¥ð╗Ðîð║ð¥ Ðüð┐ÐâÐüÐéÐÅ ð┐ð¥ð╗Ðéð¥ÐÇð░ ð│ð¥ð┤ð░ ð¢ð░ð▒ð╗ÐÄð┤ðÁð¢ð©ÐÅ ð┐ð░Ðåð©ðÁð¢Ðéð║ðÁ ð▓ð┐ðÁÐÇð▓ÐïðÁ ð▒Ðïð╗ ð▓Ðïð┐ð¥ð╗ð¢ðÁð¢ ð░ð¢ð░ð╗ð©ðÀ ð║ÐÇð¥ð▓ð© ð¢ð░┬áð░ð¢Ðéð©ð╝ð©Ðéð¥Ðàð¥ð¢ð┤ÐÇð©ð░ð╗Ðîð¢ÐïðÁ ð░ÐâÐéð¥ð░ð¢Ðéð©ÐéðÁð╗ð░ ðÉð£ðÉ-ð£2 ð©┬áð┐ð¥ÐüÐéð░ð▓ð╗ðÁð¢ ð┤ð©ð░ð│ð¢ð¥ðÀ ┬½ð┐ðÁÐÇð▓ð©Ðçð¢Ðïð╣ ð▒ð©ð╗ð©ð░ÐÇð¢Ðïð╣ Ðàð¥ð╗ð░ð¢ð│ð©Ðé┬╗; ð¢ð░ðÀð¢ð░ÐçðÁð¢ð░ ÐéðÁÐÇð░ð┐ð©ÐÅ ð┐ÐÇðÁð┐ð░ÐÇð░Ðéð░ð╝ð© ðúðöðÑðÜ, ð║ð¥Ðéð¥ÐÇð░ÐÅ ÐÅð▓ð╗ÐÅðÁÐéÐüÐÅ ÐüÐéð░ð¢ð┤ð░ÐÇÐéð¥ð╝ ð╗ðÁÐçðÁð¢ð©ÐÅ ÐìÐéð¥ð│ð¥ ðÀð░ð▒ð¥ð╗ðÁð▓ð░ð¢ð©ÐÅ.
ðÆð╝ðÁÐüÐéðÁ Ðü┬áÐéðÁð╝ ð▓┬áÐàð¥ð┤ðÁ ð┤ð©ð░ð│ð¢ð¥ÐüÐéð©ÐçðÁÐüð║ð¥ð│ð¥ ð┐ÐÇð¥ÐåðÁÐüÐüð░ ð¥ÐüÐéð░ð╗ð░ÐüÐî ð¢ðÁð┤ð¥ð¥ÐåðÁð¢ðÁð¢ð¢ð¥ð╣ ðÀð¢ð░Ðçð©ÐéðÁð╗Ðîð¢ð░ÐÅ ð░ð║Ðéð©ð▓ð¢ð¥ÐüÐéÐî Ðåð©Ðéð¥ð╗ð©ðÀð░, ð¢ðÁÐéð©ð┐ð©Ðçð¢ð░ÐÅ ð┤ð╗ÐÅ ð║ð╗ð░ÐüÐüð©ÐçðÁÐüð║ð¥ð│ð¥ ÐéðÁÐçðÁð¢ð©ÐÅ ðƒðæðÑ, ð©┬áð¢ðÁ ð▒Ðïð╗ð© ð©ÐüÐüð╗ðÁð┤ð¥ð▓ð░ð¢Ðï ð©ð╝ð╝Ðâð¢ð¥ð╗ð¥ð│ð©ÐçðÁÐüð║ð©ðÁ ð┐ð¥ð║ð░ðÀð░ÐéðÁð╗ð©, ð▓Ðüð╗ðÁð┤ÐüÐéð▓ð©ðÁ ÐçðÁð│ð¥ ð┐ð░Ðåð©ðÁð¢Ðéð║ðÁ ð¢ð░ðÀð¢ð░Ðçð░ð╗ð© ÐéðÁÐÇð░ð┐ð©ÐÄ ðƒðæðÑ ð▓┬áÐçð©ÐüÐéð¥ð╝ ð▓ð©ð┤ðÁ, ÐçÐéð¥, ð▓ðÁÐÇð¥ÐÅÐéð¢ð¥, Ðüð┐ð¥Ðüð¥ð▒ÐüÐéð▓ð¥ð▓ð░ð╗ð¥ ð┤ð░ð╗Ðîð¢ðÁð╣ÐêðÁð╝Ðâ ð┐ÐÇð¥ð│ÐÇðÁÐüÐüð©ÐÇð¥ð▓ð░ð¢ð©ÐÄ ðÀð░ð▒ð¥ð╗ðÁð▓ð░ð¢ð©ÐÅ.
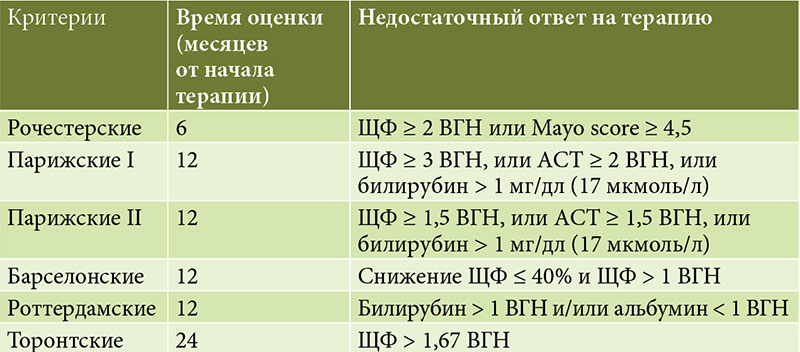
ðöÐÇÐâð│ð©ð╝ ð▓ð░ðÂð¢Ðïð╝ ð╝ð¥ð╝ðÁð¢Ðéð¥ð╝ ÐÅð▓ð╗ÐÅðÁÐéÐüÐÅ ð┐ÐÇð©ð╝ðÁð¢ðÁð¢ð©ðÁ ÐüÐéð░ð¢ð┤ð░ÐÇÐéð©ðÀð©ÐÇð¥ð▓ð░ð¢ð¢ÐïÐà ð║ÐÇð©ÐéðÁÐÇð©ðÁð▓ (ðæð░ÐÇÐüðÁð╗ð¥ð¢Ðüð║ð©Ðà, ðƒð░ÐÇð©ðÂÐüð║ð©Ðà, ðóð¥ÐÇð¥ð¢ÐéÐüð║ð©Ðà ð©┬áð┤ÐÇ.) ð┤ð╗ÐÅ ð¥ÐåðÁð¢ð║ð© ð¥Ðéð▓ðÁÐéð░ ð¢ð░┬áÐéðÁÐÇð░ð┐ð©ÐÄ (Ðéð░ð▒ð╗.┬á3): Ðàð░ÐÇð░ð║ÐéðÁÐÇ ð¥Ðéð▓ðÁÐéð░ (ð┐ð¥ð╗ð¥ðÂð©ÐéðÁð╗Ðîð¢Ðïð╣ ð©ð╗ð© ðÁð│ð¥ ð¥ÐéÐüÐâÐéÐüÐéð▓ð©ðÁ) ð┐ÐÇð©ð¥ð▒ÐÇðÁÐéð░ðÁÐé ÐÇð¥ð╗Ðî ðÀð¢ð░Ðçð©ð╝ð¥ð│ð¥ ð┤ð©ð░ð│ð¢ð¥ÐüÐéð©ÐçðÁÐüð║ð¥ð│ð¥ ð©ð¢ÐüÐéÐÇÐâð╝ðÁð¢Ðéð░, ð┐ð¥ðÀð▓ð¥ð╗ÐÅÐÅ ð║ð¥Ðüð▓ðÁð¢ð¢ð¥ ÐüÐâð┤ð©ÐéÐî ð¥┬áð║ð¥ÐÇÐÇðÁð║Ðéð¢ð¥ÐüÐéð© ð┐ðÁÐÇð▓ð¥ð¢ð░Ðçð░ð╗Ðîð¢ð¥ð╣ ð┤ð©ð░ð│ð¢ð¥ÐüÐéð©ÐçðÁÐüð║ð¥ð╣ ð│ð©ð┐ð¥ÐéðÁðÀÐï.
ðóð░ð║, Ðüð¥ð│ð╗ð░Ðüð¢ð¥ ðƒð░ÐÇð©ðÂÐüð║ð©ð╝ ð║ÐÇð©ÐéðÁÐÇð©ÐÅð╝ I, ÐâÐÇð¥ð▓ðÁð¢Ðî ð®ðñ Ðüð┐ÐâÐüÐéÐÅ 12 ð╝ðÁÐüÐÅÐåðÁð▓ ÐéðÁÐÇð░ð┐ð©ð© ð┤ð¥ð╗ðÂðÁð¢ Ðüð¥ÐüÐéð░ð▓ð╗ÐÅÐéÐî ð╝ðÁð¢ðÁðÁ ┬á3 ðÆðôðØ; Ðü┬áÐâÐçðÁÐéð¥ð╝ ðƒð░ÐÇð©ðÂÐüð║ð©Ðà ð║ÐÇð©ÐéðÁÐÇð©ðÁð▓ II ÐéÐÇðÁð▒ÐâðÁÐéÐüÐÅ Ðüð¢ð©ðÂðÁð¢ð©ðÁ ð®ðñ ð¢ð©ðÂðÁ 1,5 ðÆðôðØ. ðÆ┬áð┐ÐÇðÁð┤ÐüÐéð░ð▓ð╗ðÁð¢ð¢ð¥ð╝ ð║ð╗ð©ð¢ð©ÐçðÁÐüð║ð¥ð╝ ð¢ð░ð▒ð╗ÐÄð┤ðÁð¢ð©ð© ð┤ð©ð¢ð░ð╝ð©ð║ð░ ð®ðñ ð¢ð░┬áÐäð¥ð¢ðÁ ð╗ðÁÐçðÁð¢ð©ÐÅ ð▒Ðïð╗ð░ ð╝ð©ð¢ð©ð╝ð░ð╗Ðîð¢ð¥ð╣ (Ðüð┐ÐâÐüÐéÐÅ ÐêðÁÐüÐéÐî ð╝ðÁÐüÐÅÐåðÁð▓ ÐéðÁÐÇð░ð┐ð©ð© ð¥Ðéð╝ðÁÐçð░ð╗ð¥ÐüÐî Ðüð¢ð©ðÂðÁð¢ð©ðÁ ð®ðñ Ðü┬á412 ð┤ð¥ 312), ð▓┬áð┐ð¥Ðüð╗ðÁð┤ÐâÐÄÐëðÁð╝ ðÀð░Ðäð©ð║Ðüð©ÐÇð¥ð▓ð░ð¢ð░ ð¥ÐéÐÇð©Ðåð░ÐéðÁð╗Ðîð¢ð░ÐÅ ð┤ð©ð¢ð░ð╝ð©ð║ð░ ð▓┬áð▓ð©ð┤ðÁ ð¢ð░ÐÇð░ÐüÐéð░ð¢ð©ÐÅ Ðàð¥ð╗ðÁÐüÐéð░ðÀð░ (ð┐ð¥ð▓ÐïÐêðÁð¢ð©ðÁ ð®ðñ ð┤ð¥ 510, Ðéð¥ ðÁÐüÐéÐî ð┤ð¥ 4 ðÆðôðØ); Ðüð¢ð©ðÂðÁð¢ð©ðÁ ð╝ð░ÐÇð║ðÁÐÇð¥ð▓ Ðåð©Ðéð¥ð╗ð©ðÀð░ Ðéð░ð║ðÂðÁ ð▒Ðïð╗ð¥ ð¢ðÁð┤ð¥ÐüÐéð░Ðéð¥Ðçð¢Ðïð╝.
ð×ÐéÐüÐâÐéÐüÐéð▓ð©ðÁ ð░ð┤ðÁð║ð▓ð░Ðéð¢ð¥ð│ð¥ ð¥Ðéð▓ðÁÐéð░ ð¢ð░┬áÐéðÁÐÇð░ð┐ð©ÐÄ ðúðöðÑðÜ ð▓┬áÐüð¥ÐçðÁÐéð░ð¢ð©ð© Ðü┬áð┐ðÁÐÇÐüð©ÐüÐéð©ÐÇÐâÐÄÐëðÁð╣ ð│ð©ð┐ðÁÐÇð░ð║Ðéð©ð▓ð¢ð¥ÐüÐéÐîÐÄ Ðåð©Ðéð¥ð╗ð©Ðéð©ÐçðÁÐüð║ð©Ðà ÐäðÁÐÇð╝ðÁð¢Ðéð¥ð▓ Ðâð║ð░ðÀÐïð▓ð░ð╗ð¥ ð¢ð░┬áð▓ðÁÐÇð¥ÐÅÐéð¢ð¥ÐüÐéÐî ð▓ð░ÐÇð©ð░ð¢Ðéð¢ð¥ð│ð¥ Ðüð©ð¢ð┤ÐÇð¥ð╝ð░ ðƒðæðÑ Ðü┬áð┐ÐÇð©ðÀð¢ð░ð║ð░ð╝ð© ðÉðÿðô. ð×ð║ð¥ð¢Ðçð░ÐéðÁð╗Ðîð¢Ðïð╣ ð┤ð©ð░ð│ð¢ð¥ðÀ ð▒Ðïð╗ ÐâÐüÐéð░ð¢ð¥ð▓ð╗ðÁð¢ Ðéð¥ð╗Ðîð║ð¥ ð▓┬áÐüðÁð¢ÐéÐÅð▒ÐÇðÁ 2021 ð│.┬áð┐ÐÇð© ð¥ð▒ÐÇð░ÐëðÁð¢ð©ð© ð▓┬áð¢ð░ÐêðÁ ÐâÐçÐÇðÁðÂð┤ðÁð¢ð©ðÁ ÔÇô Ðüð┐ÐâÐüÐéÐÅ ð▒ð¥ð╗ðÁðÁ ð┤ð▓ÐâÐà ð╗ðÁÐé Ðü┬áð╝ð¥ð╝ðÁð¢Ðéð░ ð┐ðÁÐÇð▓ð©Ðçð¢ð¥ð│ð¥ ð▓ÐïÐÅð▓ð╗ðÁð¢ð©ÐÅ ð▒ð©ð¥Ðàð©ð╝ð©ÐçðÁÐüð║ð©Ðà ð╝ð░ÐÇð║ðÁÐÇð¥ð▓ ð▓ð¥Ðüð┐ð░ð╗ðÁð¢ð©ÐÅ ð©┬áÐàð¥ð╗ðÁÐüÐéð░ðÀð░.
ðƒð░Ðåð©ðÁð¢Ðéð║ðÁ ð▒Ðïð╗ð¥ ð┐ÐÇð¥ð▓ðÁð┤ðÁð¢ð¥ Ðâð│ð╗Ðâð▒ð╗ðÁð¢ð¢ð¥ðÁ ð©ð╝ð╝Ðâð¢ð¥ð╗ð¥ð│ð©ÐçðÁÐüð║ð¥ðÁ ð¥ð▒Ðüð╗ðÁð┤ð¥ð▓ð░ð¢ð©ðÁ: ð▓ÐïÐÅð▓ð╗ðÁð¢Ðï ð░ð¢Ðéð©ÐéðÁð╗ð░ ð║┬áð│ð╗ð░ð┤ð║ð¥ð╣ ð╝ÐâÐüð║Ðâð╗ð░ÐéÐâÐÇðÁ (aSMA) ð▓┬áð┤ð©ð░ð│ð¢ð¥ÐüÐéð©ÐçðÁÐüð║ð© ðÀð¢ð░Ðçð©ð╝ð¥ð╝ Ðéð©ÐéÐÇðÁ, ð░┬áÐéð░ð║ðÂðÁ ð┐ð¥ð▓ÐïÐêðÁð¢ð©ðÁ IgG, ð║ð¥Ðéð¥ÐÇÐïð╣ ÐÅð▓ð╗ÐÅðÁÐéÐüÐÅ ð▓ð░ðÂð¢Ðïð╝ ð╝ð░ÐÇð║ðÁÐÇð¥ð╝ ðÉðÿðô ð©┬áð▓Ðàð¥ð┤ð©Ðé ð▓┬áð┤ð©ð░ð│ð¢ð¥ÐüÐéð©ÐçðÁÐüð║ð©ðÁ ð║ÐÇð©ÐéðÁÐÇð©ð© ð▓ð░ÐÇð©ð░ð¢Ðéð¢ð¥ð│ð¥ Ðüð©ð¢ð┤ÐÇð¥ð╝ð░; ð┤ð╗ÐÅ ÐâÐüÐéð░ð¢ð¥ð▓ð╗ðÁð¢ð©ÐÅ ð┤ð©ð░ð│ð¢ð¥ðÀð░ ð¢ðÁð¥ð▒Ðàð¥ð┤ð©ð╝ð¥ ð¢ð░ð╗ð©Ðçð©ðÁ ð║ð░ð║ ð╝ð©ð¢ð©ð╝Ðâð╝ ð┤ð▓ÐâÐà ð║ð╗ÐÄÐçðÁð▓ÐïÐà ð║ÐÇð©ÐéðÁÐÇð©ðÁð▓ ð┤ð╗ÐÅ ð║ð░ðÂð┤ð¥ð│ð¥ ðÀð░ð▒ð¥ð╗ðÁð▓ð░ð¢ð©ÐÅ (Ðéð░ð▒ð╗.┬á2).
ðØðÁÐüð╝ð¥ÐéÐÇÐÅ ð¢ð░┬áÐäð¥ÐÇð╝ð░ð╗Ðîð¢ð¥ðÁ Ðüð¥ð¥Ðéð▓ðÁÐéÐüÐéð▓ð©ðÁ ð┐ð¥ð║ð░ðÀð░ÐéðÁð╗ðÁð╣ ð░ð¢ð░ð╗ð©ðÀð¥ð▓ ð©┬áð©ÐüÐüð╗ðÁð┤ð¥ð▓ð░ð¢ð©ð╣ ð┐ð░Ðåð©ðÁð¢Ðéð║ð© ð┤ð©ð░ð│ð¢ð¥ÐüÐéð©ÐçðÁÐüð║ð©ð╝ ð║ÐÇð©ÐéðÁÐÇð©ÐÅð╝ ðÉðÿðô, ð▓ÐïÐÅð▓ð╗ðÁð¢ð¢ð¥ðÁ Ðü┬áð┐ÐÇð©ð╝ðÁð¢ðÁð¢ð©ðÁð╝ ð¢ðÁð©ð¢ð▓ð░ðÀð©ð▓ð¢ÐïÐà ð╝ðÁÐéð¥ð┤ð¥ð▓ (ð┐ð¥ð▓ÐïÐêðÁð¢ð©ðÁ ÐéÐÇð░ð¢Ðüð░ð╝ð©ð¢ð░ðÀ ð┤ð¥ 5 ðÆðôðØ ð©┬áð¢ð░ð╗ð©Ðçð©ðÁ aSMA), ð▒Ðïð╗ð░ ð┐ÐÇð¥ð▓ðÁð┤ðÁð¢ð░ ð│ð©ÐüÐéð¥ð╗ð¥ð│ð©ÐçðÁÐüð║ð░ÐÅ ð▓ðÁÐÇð©Ðäð©ð║ð░Ðåð©ÐÅ ð┤ð©ð░ð│ð¢ð¥ðÀð░. ðÜð╗ÐÄÐçðÁð▓Ðïð╝ð© ð░ÐÇð│Ðâð╝ðÁð¢Ðéð░ð╝ð© ð▓┬áð┐ð¥ð╗ÐîðÀÐâ ð▒ð©ð¥ð┐Ðüð©ð© ÐüÐéð░ð╗ð© ð┐ð¥ð│ÐÇð░ð¢ð©Ðçð¢ÐïðÁ ðÀð¢ð░ÐçðÁð¢ð©ÐÅ ð░ð╝ð©ð¢ð¥ÐéÐÇð░ð¢ÐüÐäðÁÐÇð░ðÀ, ð║ð¥Ðéð¥ÐÇÐïðÁ ð▓┬áð┤ðÁð▒ÐÄÐéðÁ ð©┬áð¢ð░ Ðäð¥ð¢ðÁ ÐéðÁÐÇð░ð┐ð©ð© ðúðöðÑðÜ ð║ð¥ð╗ðÁð▒ð░ð╗ð©ÐüÐî ð▓┬áð┤ð©ð░ð┐ð░ðÀð¥ð¢ðÁ 4ÔÇô5 ðÆðôðØ, ð░┬áÐéð░ð║ðÂðÁ ð▓ÐïÐüð¥ð║ð░ÐÅ ð¥Ðéð▓ðÁÐéÐüÐéð▓ðÁð¢ð¢ð¥ÐüÐéÐî ð¢ð░ðÀð¢ð░ÐçðÁð¢ð©ÐÅ ð©ð╝ð╝Ðâð¢ð¥ÐüÐâð┐ÐÇðÁÐüÐüð©ð▓ð¢ð¥ð╣ ÐéðÁÐÇð░ð┐ð©ð©.
ð£ð¥ÐÇÐäð¥ð╗ð¥ð│ð©ÐçðÁÐüð║ð¥ðÁ ð©ÐüÐüð╗ðÁð┤ð¥ð▓ð░ð¢ð©ðÁ ð▒ð©ð¥ð┐Ðéð░Ðéð░ ð┐ÐÇð© ð┐ð¥ð┤ð¥ðÀÐÇðÁð¢ð©ð© ð¢ð░┬áðÉðÿðô ð©┬áð▓ð░ÐÇð©ð░ð¢Ðéð¢ÐïðÁ Ðüð©ð¢ð┤ÐÇð¥ð╝Ðï ðƒðæðÑ ÐÇðÁð║ð¥ð╝ðÁð¢ð┤ð¥ð▓ð░ð¢ð¥ ð║ð░ð║ ðÀð░ÐÇÐâð▒ðÁðÂð¢Ðïð╝ð© ð│ð░ð╣ð┤ð╗ð░ð╣ð¢ð░ð╝ð©, Ðéð░ð║ ð©┬áð¥ÐéðÁÐçðÁÐüÐéð▓ðÁð¢ð¢Ðïð╝ð© Ðìð║Ðüð┐ðÁÐÇÐéð░ð╝ð© [2, 3]. ðí┬áÐâÐçðÁÐéð¥ð╝ ð┐ð¥ÐéðÁð¢Ðåð©ð░ð╗Ðîð¢ÐïÐà ÐÇð©Ðüð║ð¥ð▓ ð┤ð╗ð©ÐéðÁð╗Ðîð¢ð¥ð╣ ð©ð╝ð╝Ðâð¢ð¥ÐüÐâð┐ÐÇðÁÐüÐüð©ð© [10] ð┤ð╗ÐÅ ð©ð¢ð©Ðåð©ð░Ðåð©ð© ðÿðíðó ÐéÐÇðÁð▒ÐâðÁÐéÐüÐÅ ð▓ÐïÐüð¥ð║ð░ÐÅ ð┤ð©ð░ð│ð¢ð¥ÐüÐéð©ÐçðÁÐüð║ð░ÐÅ ð┤ð¥ÐüÐéð¥ð▓ðÁÐÇð¢ð¥ÐüÐéÐî, ÐçÐéð¥ ð┤ðÁð╗ð░ðÁÐé ð¢ðÁð┤ð¥ð┐ÐâÐüÐéð©ð╝ð¥ð╣ ð│ð©ð┐ðÁÐÇð┤ð©ð░ð│ð¢ð¥ÐüÐéð©ð║Ðâ. ð×ð┤ð¢ð░ð║ð¥ ð▓┬áÐÇÐÅð┤ðÁ ð║ð╗ð©ð¢ð©ÐçðÁÐüð║ð©Ðà Ðüð©ÐéÐâð░Ðåð©ð╣, ð║ð¥ð│ð┤ð░ ð┤ð░ðÂðÁ ð┐ÐÇð© ð┐ð¥ÐéðÁð¢Ðåð©ð░ð╗Ðîð¢ð¥ ð▓ÐïÐüð¥ð║ð¥ð╣ ð│ð©ÐüÐéð¥ð╗ð¥ð│ð©ÐçðÁÐüð║ð¥ð╣ ð░ð║Ðéð©ð▓ð¢ð¥ÐüÐéð© ð¢ðÁ┬áð┤ð¥ÐüÐéð©ð│ð░ðÁÐéÐüÐÅ ð┤ð©ð░ð│ð¢ð¥ÐüÐéð©ÐçðÁÐüð║ð©ð╣ ð┐ð¥ÐÇð¥ð│ ð┐ð¥┬áð║ÐÇð©ÐéðÁÐÇð©ÐÅð╝ ðÉðÿðô, ð¢ðÁð╝ðÁð┤ð╗ðÁð¢ð¢ð¥ðÁ ð┐ÐÇð¥ð▓ðÁð┤ðÁð¢ð©ðÁ ð▒ð©ð¥ð┐Ðüð©ð© ð┐ðÁÐçðÁð¢ð© ð╝ð¥ðÂðÁÐé ð▒ÐïÐéÐî ð¢ðÁÐåðÁð╗ðÁÐüð¥ð¥ð▒ÐÇð░ðÀð¢Ðïð╝. ðÆ┬áÐéð░ð║ð©Ðà Ðüð╗ÐâÐçð░ÐÅÐà ð┐ÐÇðÁð┤ð┐ð¥ÐçÐéð©ÐéðÁð╗Ðîð¢ð¥ð╣ ÐÅð▓ð╗ÐÅðÁÐéÐüÐÅ ð¢ðÁð©ð¢ð▓ð░ðÀð©ð▓ð¢ð░ÐÅ ÐüÐéÐÇð░ÐéðÁð│ð©ÐÅ, ð┐ÐÇðÁð┤ð┐ð¥ð╗ð░ð│ð░ÐÄÐëð░ÐÅ ð┤ð©ð¢ð░ð╝ð©ÐçðÁÐüð║ð¥ðÁ ð¢ð░ð▒ð╗ÐÄð┤ðÁð¢ð©ðÁ Ðü┬áð▓ð¥ðÀð╝ð¥ðÂð¢ð¥ÐüÐéÐîÐÄ ð┐ðÁÐÇðÁÐüð╝ð¥ÐéÐÇð░ ð▓ð¥ð┐ÐÇð¥Ðüð░ ð¥┬áð▒ð©ð¥ð┐Ðüð©ð© ð┐ÐÇð© ð©ðÀð╝ðÁð¢ðÁð¢ð©ð© ð╗ð░ð▒ð¥ÐÇð░Ðéð¥ÐÇð¢ÐïÐà ð┐ð¥ð║ð░ðÀð░ÐéðÁð╗ðÁð╣ ð©┬áð¢ð░ÐÇð░ÐüÐéð░ð¢ð©ð© ð░ð║Ðéð©ð▓ð¢ð¥ÐüÐéð©.
ð×Ðüð¥ð▒Ðïð╣ ð¢ð░ÐâÐçð¢Ðïð╣ ð©┬áð║ð╗ð©ð¢ð©ÐçðÁÐüð║ð©ð╣ ð©ð¢ÐéðÁÐÇðÁÐü ð┐ÐÇðÁð┤ÐüÐéð░ð▓ð╗ÐÅðÁÐé ÐÇð¥ð╗Ðî Ðäð©ð▒ÐÇð¥Ðìð╗ð░ÐüÐéð¥ð╝ðÁÐéÐÇð©ð© ð▓┬áð¥ÐåðÁð¢ð║ðÁ ÐüÐéð░ð┤ð©ð© Ðäð©ð▒ÐÇð¥ðÀð░ ð┐ðÁÐçðÁð¢ð© Ðâ┬áð┐ð░Ðåð©ðÁð¢Ðéð¥ð▓ Ðü┬áð░ÐâÐéð¥ð©ð╝ð╝Ðâð¢ð¢Ðïð╝ð© ðÀð░ð▒ð¥ð╗ðÁð▓ð░ð¢ð©ÐÅð╝ð©, ð▓┬áÐçð░ÐüÐéð¢ð¥ÐüÐéð© ð┐ÐÇð© ð▓ð░ÐÇð©ð░ð¢Ðéð¢ð¥ð╝ Ðüð©ð¢ð┤ÐÇð¥ð╝ðÁ ðƒðæðÑ. ðØð░Ðê ð╝ð¢ð¥ð│ð¥ð╗ðÁÐéð¢ð©ð╣ ð║ð╗ð©ð¢ð©ÐçðÁÐüð║ð©ð╣ ð¥ð┐ÐïÐé Ðüð▓ð©ð┤ðÁÐéðÁð╗ÐîÐüÐéð▓ÐâðÁÐé ð¥┬áÐéðÁð¢ð┤ðÁð¢Ðåð©ð© ð║┬áð│ð©ð┐ðÁÐÇð┤ð©ð░ð│ð¢ð¥ÐüÐéð©ð║ðÁ Ðäð©ð▒ÐÇð¥ðÀð░ ð┐ÐÇð© ð©Ðüð┐ð¥ð╗ÐîðÀð¥ð▓ð░ð¢ð©ð© ð┤ð░ð¢ð¢ð¥ð│ð¥ ð╝ðÁÐéð¥ð┤ð░: ð║ð░ð║ ð▓┬áð┤ðÁð▒ÐÄÐéðÁ ðÀð░ð▒ð¥ð╗ðÁð▓ð░ð¢ð©ÐÅ, Ðéð░ð║ ð©┬áð¢ð░ ð¢ð░Ðçð░ð╗Ðîð¢ð¥ð╝ ÐìÐéð░ð┐ðÁ ð©ð╝ð╝Ðâð¢ð¥ÐüÐâð┐ÐÇðÁÐüÐüð©ð▓ð¢ð¥ð╣ ÐéðÁÐÇð░ð┐ð©ð© ð¢ðÁÐÇðÁð┤ð║ð¥ ÐÇðÁð│ð©ÐüÐéÐÇð©ÐÇÐâÐÄÐéÐüÐÅ ð┐ð¥ð▓ÐïÐêðÁð¢ð¢ÐïðÁ ð┐ð¥ð║ð░ðÀð░ÐéðÁð╗ð© ðÂðÁÐüÐéð║ð¥ÐüÐéð© ð┐ðÁÐçðÁð¢ð©, ð║ð¥Ðéð¥ÐÇÐïðÁ ð┤ðÁð╝ð¥ð¢ÐüÐéÐÇð©ÐÇÐâÐÄÐé ðÀð¢ð░Ðçð©ÐéðÁð╗Ðîð¢ð¥ðÁ Ðüð¢ð©ðÂðÁð¢ð©ðÁ ð▓┬áð┤ð©ð¢ð░ð╝ð©ð║ðÁ. ðíð¥ð▓ÐÇðÁð╝ðÁð¢ð¢ÐïðÁ ð┐ÐÇðÁð┤ÐüÐéð░ð▓ð╗ðÁð¢ð©ÐÅ ð¥┬áð╝ðÁÐàð░ð¢ð©ðÀð╝ð░Ðà Ðäð©ð▒ÐÇð¥ð│ðÁð¢ðÁðÀð░ ð©┬áð╝ð¥ÐÇÐäð¥ð╗ð¥ð│ð©ÐçðÁÐüð║ð©Ðà ð¥Ðüð¥ð▒ðÁð¢ð¢ð¥ÐüÐéÐÅÐà ð┐ðÁÐçðÁð¢ð© ð┐ÐÇð© ðÉðÿðùðƒ ð┐ð¥ðÀð▓ð¥ð╗ÐÅÐÄÐé ð┐ÐÇðÁð┤ð┐ð¥ð╗ð¥ðÂð©ÐéÐî, ÐçÐéð¥ ð┐ð¥ð▓ÐïÐêðÁð¢ð¢ÐïðÁ ðÀð¢ð░ÐçðÁð¢ð©ÐÅ ðÂðÁÐüÐéð║ð¥ÐüÐéð© ð¥ð▒ÐâÐüð╗ð¥ð▓ð╗ðÁð¢Ðï ð¢ðÁ┬áÐéð¥ð╗Ðîð║ð¥ Ðüð¥ð▒ÐüÐéð▓ðÁð¢ð¢ð¥ Ðäð©ð▒ÐÇð¥ðÀð¥ð╝, ð¢ð¥┬áð© ð░ð║Ðéð©ð▓ð¢ð¥ÐüÐéÐîÐÄ ð░ÐâÐéð¥ð©ð╝ð╝Ðâð¢ð¢ð¥ð│ð¥ ð▓ð¥Ðüð┐ð░ð╗ðÁð¢ð©ÐÅ, ð┤ð░ðÂðÁ ð┐ÐÇð© ÐâÐÇð¥ð▓ð¢ðÁ ÐéÐÇð░ð¢Ðüð░ð╝ð©ð¢ð░ðÀ, ð¢ðÁ┬áð┐ÐÇðÁð▓ÐïÐêð░ÐÄÐëðÁð╝ 5 ðÆðôðØ (Ðäð¥ÐÇð╝ð░ð╗Ðîð¢ð¥ ð┤ð¥ð┐ÐâÐüÐéð©ð╝Ðïð╣ ð┐ð¥ÐÇð¥ð│ ð┤ð╗ÐÅ ð┐ÐÇð¥ð▓ðÁð┤ðÁð¢ð©ÐÅ ð©ÐüÐüð╗ðÁð┤ð¥ð▓ð░ð¢ð©ÐÅ). ðƒÐÇðÁð┤ÐüÐéð░ð▓ð╗ðÁð¢ð¢Ðïð╣ ð║ð╗ð©ð¢ð©ÐçðÁÐüð║ð©ð╣ Ðüð╗ÐâÐçð░ð╣ ð¢ð░ð│ð╗ÐÅð┤ð¢ð¥ ð┤ðÁð╝ð¥ð¢ÐüÐéÐÇð©ÐÇÐâðÁÐé ÐÇð░ÐüÐàð¥ðÂð┤ðÁð¢ð©ðÁ ð╝ðÁðÂð┤Ðâ ð┤ð░ð¢ð¢Ðïð╝ð© ð¢ðÁð©ð¢ð▓ð░ðÀð©ð▓ð¢ð¥ð╣ ð©┬áð╝ð¥ÐÇÐäð¥ð╗ð¥ð│ð©ÐçðÁÐüð║ð¥ð╣ ð¥ÐåðÁð¢ð║ð©: ð┐ÐÇð© ð┐ð¥ð║ð░ðÀð░ÐéðÁð╗ðÁ ðÂðÁÐüÐéð║ð¥ÐüÐéð© ð┐ðÁÐçðÁð¢ð© 11,7 ð║ðƒð░ (F3 ð┐ð¥┬áÐêð║ð░ð╗ðÁ METAVIR) ð│ð©ÐüÐéð¥ð╗ð¥ð│ð©ÐçðÁÐüð║ð© ð▒Ðïð╗ð░ ð▓ðÁÐÇð©Ðäð©Ðåð©ÐÇð¥ð▓ð░ð¢ð░ ÐüÐéð░ð┤ð©ÐÅ F2, ÐçÐéð¥ ð┐ð¥ð┤Ðéð▓ðÁÐÇðÂð┤ð░ðÁÐé ð¥ð│ÐÇð░ð¢ð©ÐçðÁð¢ð©ÐÅ ð┐ÐÇð©ð╝ðÁð¢ðÁð¢ð©ÐÅ Ðìð╗ð░ÐüÐéð¥ð╝ðÁÐéÐÇð©ð© ð▓┬áÐâÐüð╗ð¥ð▓ð©ÐÅÐà ð░ð║Ðéð©ð▓ð¢ð¥ð│ð¥ ð▓ð¥Ðüð┐ð░ð╗ðÁð¢ð©ÐÅ.
ðíð╗ðÁð┤ÐâðÁÐé ð¥Ðéð╝ðÁÐéð©ÐéÐî, ÐçÐéð¥ ÐÇðÁðÀÐâð╗ÐîÐéð░ÐéÐï ð¥ð┐Ðâð▒ð╗ð©ð║ð¥ð▓ð░ð¢ð¢ÐïÐà ð©ÐüÐüð╗ðÁð┤ð¥ð▓ð░ð¢ð©ð╣ ð┐ð¥┬áÐìÐéð¥ð╝Ðâ ð▓ð¥ð┐ÐÇð¥ÐüÐâ ð¥Ðéð╗ð©Ðçð░ÐÄÐéÐüÐÅ ð¢ðÁð¥ð┤ð¢ð¥ðÀð¢ð░Ðçð¢ð¥ÐüÐéÐîÐÄ. ðáÐÅð┤ ð░ð▓Ðéð¥ÐÇð¥ð▓ Ðâð║ð░ðÀÐïð▓ð░ðÁÐé ð¢ð░┬áð¢ðÁð¥ð▒Ðàð¥ð┤ð©ð╝ð¥ÐüÐéÐî ð║ð¥ÐÇÐÇðÁð║Ðéð©ÐÇð¥ð▓ð║ð© ð┤ð©ð░ð│ð¢ð¥ÐüÐéð©ÐçðÁÐüð║ð©Ðà ð┐ð¥ÐÇð¥ð│ð¥ð▓ ð▓┬áÐüÐéð¥ÐÇð¥ð¢Ðâ ð┐ð¥ð▓ÐïÐêðÁð¢ð©ÐÅ ð┤ð╗ÐÅ ð┐ð░Ðåð©ðÁð¢Ðéð¥ð▓ Ðü┬áðƒðæðÑ. ðÆ┬áÐçð░ÐüÐéð¢ð¥ÐüÐéð©, ð▓┬áð©ÐüÐüð╗ðÁð┤ð¥ð▓ð░ð¢ð©ð© C. Corpechot ð©┬áÐüð¥ð░ð▓Ðé. ð¥ð┐Ðéð©ð╝ð░ð╗Ðîð¢Ðïð╝ ð┐ð¥ÐÇð¥ð│ð¥ð▓Ðïð╝ ðÀð¢ð░ÐçðÁð¢ð©ðÁð╝ ðÂðÁÐüÐéð║ð¥ÐüÐéð© ð┐ðÁÐçðÁð¢ð© ð┤ð╗ÐÅ ð┤ð©ð░ð│ð¢ð¥ÐüÐéð©ð║ð© Ðåð©ÐÇÐÇð¥ðÀð░ Ðâ┬áð┐ð░Ðåð©ðÁð¢Ðéð¥ð▓ Ðü┬áðƒðæðÑ ð©┬áðƒðíðÑ ð▒Ðïð╗ ð┐ÐÇðÁð┤ð╗ð¥ðÂðÁð¢ ÐâÐÇð¥ð▓ðÁð¢Ðî 17,3 ð║ðƒð░ [11]. ðÆ┬áÐéð¥ ðÂðÁ ð▓ÐÇðÁð╝ÐÅ ÐÇðÁðÀÐâð╗ÐîÐéð░ÐéÐï ð¢ðÁð┤ð░ð▓ð¢ð¥ ð┐ÐÇð¥ð▓ðÁð┤ðÁð¢ð¢ð¥ð│ð¥ ð╝ðÁÐéð░ð░ð¢ð░ð╗ð©ðÀð░ 2023 ð│., ð▓ð║ð╗ÐÄÐçð©ð▓ÐêðÁð│ð¥ 60 ð©ÐüÐüð╗ðÁð┤ð¥ð▓ð░ð¢ð©ð╣, ð┤ðÁð╝ð¥ð¢ÐüÐéÐÇð©ÐÇÐâÐÄÐé ð▓ÐïÐüð¥ð║ÐâÐÄ ð┤ð©ð░ð│ð¢ð¥ÐüÐéð©ÐçðÁÐüð║ÐâÐÄ Ðéð¥Ðçð¢ð¥ÐüÐéÐî Ðäð©ð▒ÐÇð¥Ðìð╗ð░ÐüÐéð¥ð╝ðÁÐéÐÇð©ð© ð┐ÐÇð© ð▓ð░ÐÇð©ð░ð¢Ðéð¢ÐïÐà Ðüð©ð¢ð┤ÐÇð¥ð╝ð░Ðà. ðÆð╝ðÁÐüÐéðÁ Ðü┬áÐéðÁð╝ ð░ð▓Ðéð¥ÐÇÐï ð╝ðÁÐéð░ð░ð¢ð░ð╗ð©ðÀð░ Ðüð┐ÐÇð░ð▓ðÁð┤ð╗ð©ð▓ð¥ ð¥Ðéð╝ðÁÐçð░ÐÄÐé, ÐçÐéð¥ ð▓ð▓ð©ð┤Ðâ ð¢ðÁð┤ð¥ÐüÐéð░Ðéð¥Ðçð¢ð¥ÐüÐéð© ð┤ð░ð¢ð¢ÐïÐà ð▓ð¥ð┐ÐÇð¥Ðü ð¥┬áð▓ð╗ð©ÐÅð¢ð©ð© ÐâÐÇð¥ð▓ð¢ÐÅ ðÉðøðó ð¢ð░┬áÐÇð░ðÀð╗ð©Ðçð©ÐÅ ð▓┬áð┤ð©ð░ð│ð¢ð¥ÐüÐéð©ÐçðÁÐüð║ð¥ð╣ Ðéð¥Ðçð¢ð¥ÐüÐéð© ð╝ðÁðÂð┤Ðâ ð┐ð░Ðåð©ðÁð¢Ðéð░ð╝ð© Ðü┬áðÉðÿðô ð©┬áðƒðæðÑ ð¥ÐüÐéð░ðÁÐéÐüÐÅ ð¥Ðéð║ÐÇÐïÐéÐïð╝ [12].
ðöð░ð╗Ðîð¢ðÁð╣ÐêðÁðÁ ð┤ð©ð¢ð░ð╝ð©ÐçðÁÐüð║ð¥ðÁ ð¢ð░ð▒ð╗ÐÄð┤ðÁð¢ð©ðÁ ðÀð░ ð┐ð░Ðåð©ðÁð¢Ðéð║ð¥ð╣ ð┐ÐÇð¥ð┤ðÁð╝ð¥ð¢ÐüÐéÐÇð©ÐÇð¥ð▓ð░ð╗ð¥ ÐÇðÁð│ÐÇðÁÐüÐü Ðäð©ð▒ÐÇð¥ðÀð░ ð┐ðÁÐçðÁð¢ð© ð¢ð░┬áÐäð¥ð¢ðÁ ðÿðíðó (ð┐ÐÇð© ð┐ðÁÐÇð▓ð©Ðçð¢ð¥ð╝ ð¥ð▒Ðüð╗ðÁð┤ð¥ð▓ð░ð¢ð©ð© ð│ð©ÐüÐéð¥ð╗ð¥ð│ð©ÐçðÁÐüð║ð© ð¥ð┐ÐÇðÁð┤ðÁð╗ÐÅð╗ð░ÐüÐî ÐüÐéð░ð┤ð©ÐÅ F2, ð┐ÐÇð© ð║ð¥ð¢ÐéÐÇð¥ð╗Ðîð¢ð¥ð╝ ð¥ð▒Ðüð╗ðÁð┤ð¥ð▓ð░ð¢ð©ð© Ðüð┐ÐâÐüÐéÐÅ ÐéÐÇð© ð│ð¥ð┤ð░ ð▒Ðïð╗ð░ ð▓ðÁÐÇð©Ðäð©Ðåð©ÐÇð¥ð▓ð░ð¢ð░ ÐüÐéð░ð┤ð©ÐÅ F1), ÐçÐéð¥ Ðüð¥ð¥Ðéð▓ðÁÐéÐüÐéð▓ÐâðÁÐé Ðüð¥ð▓ÐÇðÁð╝ðÁð¢ð¢Ðïð╝ ð¢ð░ÐâÐçð¢Ðïð╝ ð┤ð░ð¢ð¢Ðïð╝, Ðéð░ð║ ð║ð░ð║┬á ð▓ð¥ðÀð╝ð¥ðÂð¢ð¥ÐüÐéÐî ð¥ð▒ÐÇð░Ðéð¢ð¥ð│ð¥ ÐÇð░ðÀð▓ð©Ðéð©ÐÅ Ðäð©ð▒ÐÇð¥ðÀð░ ð┐ðÁÐçðÁð¢ð© ð┐ð¥ð┤Ðéð▓ðÁÐÇðÂð┤ðÁð¢ð░ ð╝ð¢ð¥ð│ð©ð╝ð© ð░ð▓Ðéð¥ÐÇð░ð╝ð© [13, 14].
ðíð¥ÐàÐÇð░ð¢ÐÅðÁÐéÐüÐÅ ð¢ðÁð║ð¥Ðéð¥ÐÇð░ÐÅ ð¢ðÁð¥ð┐ÐÇðÁð┤ðÁð╗ðÁð¢ð¢ð¥ÐüÐéÐî ð▓┬áð¥Ðéð¢ð¥ÐêðÁð¢ð©ð© ð┐ÐÇð¥ð┤ð¥ð╗ðÂð©ÐéðÁð╗Ðîð¢ð¥ÐüÐéð© ð╗ðÁÐçðÁð¢ð©ÐÅ ð©┬áð┐ÐÇð¥Ðéð¥ð║ð¥ð╗ð¥ð▓ ð┐ÐÇðÁð║ÐÇð░ÐëðÁð¢ð©ÐÅ ð©ð╝ð╝Ðâð¢ð¥ÐüÐâð┐ÐÇðÁÐüÐüð©ð▓ð¢ð¥ð╣ ÐéðÁÐÇð░ð┐ð©ð© ð┐ÐÇð© ð▓ð░ÐÇð©ð░ð¢Ðéð¢ÐïÐà Ðüð©ð¢ð┤ÐÇð¥ð╝ð░Ðà; ð┐ÐÇð© ð║ð╗ð░ÐüÐüð©ÐçðÁÐüð║ð¥ð╝ ðÉðÿðô ð¥Ðéð╝ðÁð¢ð░ ðÿðíðó ÐÇðÁð║ð¥ð╝ðÁð¢ð┤ÐâðÁÐéÐüÐÅ ð┐ÐÇð© ð┤ð¥ÐüÐéð©ðÂðÁð¢ð©ð© ð©ð¢ð┤ðÁð║Ðüð░ ð│ð©ÐüÐéð¥ð╗ð¥ð│ð©ÐçðÁÐüð║ð¥ð╣ ð░ð║Ðéð©ð▓ð¢ð¥ÐüÐéð© (ðÿðôðÉ) ð╝ðÁð¢ðÁðÁ 4 ð▒ð░ð╗ð╗ð¥ð▓ [2]. ðÆ┬áð┐ÐÇðÁð┤ÐüÐéð░ð▓ð╗ðÁð¢ð¢ð¥ð╝ ð║ð╗ð©ð¢ð©ÐçðÁÐüð║ð¥ð╝ Ðüð╗ÐâÐçð░ðÁ ð¢ð░┬áð¥Ðüð¢ð¥ð▓ð░ð¢ð©ð© ð┤ð╗ð©ÐéðÁð╗Ðîð¢ð¥ð│ð¥ ð┐ðÁÐÇð©ð¥ð┤ð░ ð╗ðÁÐçðÁð¢ð©ÐÅ, ð┐ð¥ð│ÐÇð░ð¢ð©Ðçð¢ÐïÐà ðÀð¢ð░ÐçðÁð¢ð©ð╣ ðÿðôðÉ ð©┬áÐÇð░ðÀð▓ð©Ðéð©ÐÅ ð¢ðÁðÂðÁð╗ð░ÐéðÁð╗Ðîð¢ð¥ð│ð¥ ÐÅð▓ð╗ðÁð¢ð©ÐÅ (ð¥ÐüÐéðÁð¥ð┐ð¥ÐÇð¥ðÀð░) ð▒Ðïð╗ð░ ð©ðÀð▒ÐÇð░ð¢ð░ ð©ð¢ð┤ð©ð▓ð©ð┤Ðâð░ð╗Ðîð¢ð░ÐÅ Ðéð░ð║Ðéð©ð║ð░: ð┐ÐÇð¥ð▓ðÁð┤ðÁð¢ð░ ð┐ð¥ÐüÐéðÁð┐ðÁð¢ð¢ð░ÐÅ ð¥Ðéð╝ðÁð¢ð░ ð╝ðÁÐéð©ð╗ð┐ÐÇðÁð┤ð¢ð©ðÀð¥ð╗ð¥ð¢ð░ Ðü┬áð┐ÐÇð¥ð╗ð¥ð¢ð│ð░Ðåð©ðÁð╣ ð┐ÐÇð©ðÁð╝ð░ ð░ðÀð░Ðéð©ð¥ð┐ÐÇð©ð¢ð░ ð¢ð░┬áÐêðÁÐüÐéÐî ð╝ðÁÐüÐÅÐåðÁð▓ Ðü┬áð┐ð¥Ðüð╗ðÁð┤ÐâÐÄÐëðÁð╣ ðÁð│ð¥ ð¥Ðéð╝ðÁð¢ð¥ð╣.
ðöð©ð¢ð░ð╝ð©ÐçðÁÐüð║ð¥ðÁ ð¢ð░ð▒ð╗ÐÄð┤ðÁð¢ð©ðÁ ð©┬áð╗ð░ð▒ð¥ÐÇð░Ðéð¥ÐÇð¢Ðïð╣ ð╝ð¥ð¢ð©Ðéð¥ÐÇð©ð¢ð│ ð┐ð¥ð┤Ðéð▓ðÁÐÇð┤ð©ð╗ð© ÐìÐäÐäðÁð║Ðéð©ð▓ð¢ð¥ÐüÐéÐî ð▓Ðïð▒ÐÇð░ð¢ð¢ð¥ð│ð¥ ð┐ð¥ð┤Ðàð¥ð┤ð░: ð▒ð©ð¥Ðàð©ð╝ð©ÐçðÁÐüð║ð©ðÁ ð┐ð¥ð║ð░ðÀð░ÐéðÁð╗ð© ð¥ÐüÐéð░ð▓ð░ð╗ð©ÐüÐî ð▓┬áð┐ÐÇðÁð┤ðÁð╗ð░Ðà ð¢ð¥ÐÇð╝Ðï ð┐ÐÇð© ð║ð¥ð¢ÐéÐÇð¥ð╗Ðîð¢ð¥ð╝ ð¥ð▒Ðüð╗ðÁð┤ð¥ð▓ð░ð¢ð©ð© ÐçðÁÐÇðÁðÀ ÐéÐÇð© ð╝ðÁÐüÐÅÐåð░ ð┐ð¥Ðüð╗ðÁ ð¥Ðéð╝ðÁð¢Ðï ð╝ðÁÐéð©ð╗ð┐ÐÇðÁð┤ð¢ð©ðÀð¥ð╗ð¥ð¢ð░, ð░┬áÐéð░ð║ðÂðÁ ÐçðÁÐÇðÁðÀ ð▓ð¥ÐüðÁð╝Ðî ð╝ðÁÐüÐÅÐåðÁð▓ ð©┬áð┤ð¥ ð¢ð░ÐüÐéð¥ÐÅÐëðÁð│ð¥ ð▓ÐÇðÁð╝ðÁð¢ð© ð┐ð¥Ðüð╗ðÁ ð┐ð¥ð╗ð¢ð¥ð╣ ð¥Ðéð╝ðÁð¢Ðï ð░ðÀð░Ðéð©ð¥ð┐ÐÇð©ð¢ð░, ÐçÐéð¥ Ðüð▓ð©ð┤ðÁÐéðÁð╗ÐîÐüÐéð▓ÐâðÁÐé ð¥┬áÐüÐéð¥ð╣ð║ð¥ð╣ ð▒ð©ð¥Ðàð©ð╝ð©ÐçðÁÐüð║ð¥ð╣ ÐÇðÁð╝ð©ÐüÐüð©ð©.
ðíð╗ðÁð┤ÐâðÁÐé ð¥Ðéð╝ðÁÐéð©ÐéÐî, ÐçÐéð¥ ð║ÐÇð©ÐéðÁÐÇð©ð© ð¥Ðéð╝ðÁð¢Ðï ðÿðíðó ð┐ÐÇð© ð▓ð░ÐÇð©ð░ð¢Ðéð¢ð¥ð╝ Ðüð©ð¢ð┤ÐÇð¥ð╝ðÁ ðƒðæðÑ Ðü┬áð┐ÐÇð©ðÀð¢ð░ð║ð░ð╝ð© ðÉðÿðô ð¢ðÁ┬áÐüÐéð░ð¢ð┤ð░ÐÇÐéð©ðÀð©ÐÇð¥ð▓ð░ð¢Ðï ð▓┬áÐüð▓ÐÅðÀð© Ðü┬áð│ðÁÐéðÁÐÇð¥ð│ðÁð¢ð¢ð¥ÐüÐéÐîÐÄ ð║ð╗ð©ð¢ð©ÐçðÁÐüð║ð©Ðà ð┐ÐÇð¥ÐÅð▓ð╗ðÁð¢ð©ð╣, ð┐ÐÇð© ð║ð¥Ðéð¥ÐÇÐïÐà ð╝ð¥ð│ÐâÐé ð┤ð¥ð╝ð©ð¢ð©ÐÇð¥ð▓ð░ÐéÐî ð┐ÐÇð©ðÀð¢ð░ð║ð© ð╗ð©ð▒ð¥ ðƒðæðÑ, ð╗ð©ð▒ð¥ ðÉðÿðô, ð╗ð©ð▒ð¥ ð┐ÐÇð©ÐüÐâÐéÐüÐéð▓ÐâðÁÐé ð©Ðà ÐÇð░ð▓ð¢ð¥ðÀð¢ð░Ðçð¢ð¥ðÁ Ðüð¥ÐçðÁÐéð░ð¢ð©ðÁ [3]. ðÆ┬áÐâÐüð╗ð¥ð▓ð©ÐÅÐà ð¥ÐéÐüÐâÐéÐüÐéð▓ð©ÐÅ Ðâð¢ð©Ðäð©Ðåð©ÐÇð¥ð▓ð░ð¢ð¢ÐïÐà ÐÇðÁð║ð¥ð╝ðÁð¢ð┤ð░Ðåð©ð╣ ð┐ð¥┬áÐéðÁÐÇð░ð┐ð©ð© ð©┬áðÁðÁ ð┐ÐÇðÁð║ÐÇð░ÐëðÁð¢ð©ÐÄ ð┐ÐÇð© ð▓ð░ÐÇð©ð░ð¢Ðéð¢ÐïÐà Ðüð©ð¢ð┤ÐÇð¥ð╝ð░Ðà ð┐ÐÇðÁð┤ÐüÐéð░ð▓ð╗ÐÅðÁÐéÐüÐÅ ÐåðÁð╗ðÁÐüð¥ð¥ð▒ÐÇð░ðÀð¢Ðïð╝ ð┐ÐÇð©ð╝ðÁð¢ðÁð¢ð©ðÁ ð┐ÐÇð©ð¢Ðåð©ð┐ð¥ð▓, ð┐ÐÇð©ð¢ÐÅÐéÐïÐà ð┤ð╗ÐÅ ðÉðÿðô, ð░┬áð©ð╝ðÁð¢ð¢ð¥ ÐÇð░ÐüÐüð╝ð¥ÐéÐÇðÁð¢ð©ðÁ ð▓ð¥ð┐ÐÇð¥Ðüð░ ð¥ð▒┬áð¥Ðéð╝ðÁð¢ðÁ ðÿðíðó ÐçðÁÐÇðÁðÀ ð┤ð▓ð░ ð│ð¥ð┤ð░ ð╗ðÁÐçðÁð¢ð©ÐÅ ð┐ÐÇð© ÐâÐüð╗ð¥ð▓ð©ð© ð┤ð¥ÐüÐéð©ðÂðÁð¢ð©ÐÅ ð│ð©ÐüÐéð¥ð╗ð¥ð│ð©ÐçðÁÐüð║ð¥ð╣ ÐÇðÁð╝ð©ÐüÐüð©ð©.
ðùð░ð║ð╗ÐÄÐçðÁð¢ð©ðÁ
ðÆ┬áð┐ÐÇðÁð┤ÐüÐéð░ð▓ð╗ðÁð¢ð¢ð¥ð╝ ð║ð╗ð©ð¢ð©ÐçðÁÐüð║ð¥ð╝ Ðüð╗ÐâÐçð░ðÁ ð┤ð¥ð╝ð©ð¢ð©ÐÇÐâÐÄÐëð©ð╝ ðÀð░ð▒ð¥ð╗ðÁð▓ð░ð¢ð©ðÁð╝ ÐÅð▓ð╗ÐÅð╗ÐüÐÅ ðƒðæðÑ, ÐçÐéð¥ ð┐ð¥ð┤Ðéð▓ðÁÐÇðÂð┤ð░ð╗ð¥ÐüÐî ð¢ð░ð╗ð©Ðçð©ðÁð╝ ð║ð╗ð©ð¢ð©ð║ð¥-ð▒ð©ð¥Ðàð©ð╝ð©ÐçðÁÐüð║ð¥ð│ð¥ Ðàð¥ð╗ðÁÐüÐéð░ðÀð░; ð¥┬áðÀð¢ð░Ðçð©ÐéðÁð╗Ðîð¢ð¥ð╝ ð▓ð║ð╗ð░ð┤ðÁ ðÉðÿðô Ðüð▓ð©ð┤ðÁÐéðÁð╗ÐîÐüÐéð▓ð¥ð▓ð░ð╗ð¥ ð▒ÐïÐüÐéÐÇð¥ðÁ ð┐ÐÇð¥ð│ÐÇðÁÐüÐüð©ÐÇð¥ð▓ð░ð¢ð©ðÁ Ðäð©ð▒ÐÇð¥ðÀð░ ð┐ðÁÐçðÁð¢ð©.
ðíð╗ðÁð┤ÐâðÁÐé ð¥Ðéð╝ðÁÐéð©ÐéÐî, ÐçÐéð¥ ð▓ð░ÐÇð©ð░ð¢Ðéð¢ÐïðÁ Ðüð©ð¢ð┤ÐÇð¥ð╝Ðï Ðàð░ÐÇð░ð║ÐéðÁÐÇð©ðÀÐâÐÄÐéÐüÐÅ ðÀð¢ð░Ðçð©ÐéðÁð╗Ðîð¢Ðïð╝ ð┐ð¥ð╗ð©ð╝ð¥ÐÇÐäð©ðÀð╝ð¥ð╝ ð║ð╗ð©ð¢ð©ÐçðÁÐüð║ð©Ðà, ð╗ð░ð▒ð¥ÐÇð░Ðéð¥ÐÇð¢ÐïÐà ð©┬áð│ð©ÐüÐéð¥ð╗ð¥ð│ð©ÐçðÁÐüð║ð©Ðà ð┐ÐÇð©ðÀð¢ð░ð║ð¥ð▓. ðÆ┬áð¢ð░ÐâÐçð¢ð¥ð╝ Ðüð¥ð¥ð▒ÐëðÁÐüÐéð▓ðÁ ð┐ÐÇð¥ð┤ð¥ð╗ðÂð░ðÁÐéÐüÐÅ ð┤ð©Ðüð║ÐâÐüÐüð©ÐÅ ð¥Ðéð¢ð¥Ðüð©ÐéðÁð╗Ðîð¢ð¥ ð¢ð¥ðÀð¥ð╗ð¥ð│ð©ÐçðÁÐüð║ð¥ð│ð¥ ÐüÐéð░ÐéÐâÐüð░ ð┐ðÁÐÇðÁð║ÐÇðÁÐüÐéð¢ÐïÐà Ðüð©ð¢ð┤ÐÇð¥ð╝ð¥ð▓: ÐÇð░ÐüÐüð╝ð░ÐéÐÇð©ð▓ð░ÐéÐî ð╗ð© ð©Ðà ð║ð░ð║ Ðüð░ð╝ð¥ÐüÐéð¥ÐÅÐéðÁð╗Ðîð¢ÐïðÁ ð¢ð¥ðÀð¥ð╗ð¥ð│ð©ÐçðÁÐüð║ð©ðÁ ðÁð┤ð©ð¢ð©ÐåÐï ð©ð╗ð© ð║ð░ð║ ð┐ðÁÐÇðÁÐàð¥ð┤ð¢ÐïðÁ Ðäð¥ÐÇð╝Ðï ð╝ðÁðÂð┤Ðâ ð¥Ðéð┤ðÁð╗Ðîð¢Ðïð╝ð© ðÉðÿðùðƒ.
ðÉð║ÐéÐâð░ð╗Ðîð¢ð¥ÐüÐéÐî ð┐ÐÇð¥ð▒ð╗ðÁð╝Ðï ð┐ð¥ð┤ÐçðÁÐÇð║ð©ð▓ð░ðÁÐéÐüÐÅ ÐÇð¥ÐüÐéð¥ð╝ ÐÇð░Ðüð┐ÐÇð¥ÐüÐéÐÇð░ð¢ðÁð¢ð¢ð¥ÐüÐéð© ð░ÐâÐéð¥ð©ð╝ð╝Ðâð¢ð¢ÐïÐà ð┐ðÁÐÇðÁð║ÐÇðÁÐüÐéð¢ÐïÐà Ðüð©ð¢ð┤ÐÇð¥ð╝ð¥ð▓ ÐüÐÇðÁð┤ð© ð╗ð©Ðå ð╝ð¥ð╗ð¥ð┤ð¥ð│ð¥ ð©┬áÐüÐÇðÁð┤ð¢ðÁð│ð¥ ð▓ð¥ðÀÐÇð░ÐüÐéð░, ð©Ðà ð░ð│ÐÇðÁÐüÐüð©ð▓ð¢Ðïð╝ ÐéðÁÐçðÁð¢ð©ðÁð╝ Ðü┬áð▒ÐïÐüÐéÐÇÐïð╝ ÐÇð░ðÀð▓ð©Ðéð©ðÁð╝ Ðäð©ð▒ÐÇð¥ðÀð░ ð©┬áÐåð©ÐÇÐÇð¥ðÀð░ ð┐ðÁÐçðÁð¢ð©, ð░┬áÐéð░ð║ðÂðÁ ð¥ÐéÐüÐâÐéÐüÐéð▓ð©ðÁð╝ Ðâð¢ð©Ðäð©Ðåð©ÐÇð¥ð▓ð░ð¢ð¢ÐïÐà ð┐ð¥ð┤Ðàð¥ð┤ð¥ð▓ ð║┬áð┤ð©ð░ð│ð¢ð¥ÐüÐéð©ð║ðÁ ð©┬áÐéðÁÐÇð░ð┐ð©ð©. ðÆÐüðÁ ð▓ÐïÐêðÁð©ðÀð╗ð¥ðÂðÁð¢ð¢ð¥ðÁ ð¥ð┐ÐÇðÁð┤ðÁð╗ÐÅðÁÐé ð¢ðÁð¥ð▒Ðàð¥ð┤ð©ð╝ð¥ÐüÐéÐî ð┤ð░ð╗Ðîð¢ðÁð╣ÐêðÁð│ð¥ Ðâð│ð╗Ðâð▒ð╗ðÁð¢ð¢ð¥ð│ð¥ ð©ðÀÐâÐçðÁð¢ð©ÐÅ ð┤ð░ð¢ð¢ð¥ð╣ ð║ð░ÐéðÁð│ð¥ÐÇð©ð© ðÀð░ð▒ð¥ð╗ðÁð▓ð░ð¢ð©ð╣.
ðÉð▓Ðéð¥ÐÇÐï ðÀð░ÐÅð▓ð╗ÐÅÐÄÐé, ÐçÐéð¥ ð┤ð░ð¢ð¢ð░ÐÅ ÐÇð░ð▒ð¥Ðéð░, ðÁðÁ ÐéðÁð╝ð░, ð┐ÐÇðÁð┤ð╝ðÁÐé ð©┬áÐüð¥ð┤ðÁÐÇðÂð░ð¢ð©ðÁ ð¢ðÁ┬áðÀð░ÐéÐÇð░ð│ð©ð▓ð░ÐÄÐé ð║ð¥ð¢ð║ÐâÐÇð©ÐÇÐâÐÄÐëð©Ðà ð©ð¢ÐéðÁÐÇðÁÐüð¥ð▓.
ðáð░ð▒ð¥Ðéð░ ð▓Ðïð┐ð¥ð╗ð¢ðÁð¢ð░ ð▓┬áÐÇð░ð╝ð║ð░Ðà ðØðÿðá ðöðùð£.ðáðÁð│ð©ÐüÐéÐÇð░Ðåð©ð¥ð¢ð¢Ðïð╣ ð¢ð¥ð╝ðÁÐÇ ðØðÿð×ðÜðóðá: 123040700014-4.
A.V. Anisonyan, PhD, E.A. Sokolova, E.S. Sbikina PhD, E.V. Vinnitskaya, PhD, Prof., T.Yu. Khaimenova, PhD, Yu.G. Sandler, PhD, S.G. Khomeriki, PhD, Prof.
A.S. Loginov Moscow Clinical Scientific Center
Contact person: Anastasia V. Anisonyan, Anastasiya7651@yandex.ru
Autoimmune hepatitis (AIH) and primary biliary cholangitis (PBC) are immune-mediated liver diseases with different target cells ÔÇô hepatocytes and cholangiocytes respectively. Variant syndromes are characterized by a combination of clinical, laboratory, and morphological features of both diseases, which creates significant diagnostic challenges. We present a case of variant syndrome ┬½primary biliary cholangitis with features of autoimmune hepatitis┬╗. The diagnostic process spanned over two years from the initial detection of liver biochemistry abnormalities. During this period, ursodeoxycholic acid monotherapy proved insufficient, with disease progression to fibrosis stage F2 documented. Definitive diagnosis required comprehensive immunological and histological evaluation, which confirmed features of both diseases. This case highlights the critical importance of thorough investigation upon detecting liver enzyme abnormalities to determine their underlying etiology. Precise characterization of the pathological process is essential for selecting appropriate treatment. In this patient, establishing the correct diagnosis enabled timely immunosuppressive therapy, resulting in sustained biochemical and histological remission with regression of fibrosis to stage F1.
ðúð▓ð░ðÂð░ðÁð╝Ðïð╣ ð┐ð¥ÐüðÁÐéð©ÐéðÁð╗Ðî uMEDp!
ðúð▓ðÁð┤ð¥ð╝ð╗ÐÅðÁð╝ ðÆð░Ðü ð¥ Ðéð¥ð╝, ÐçÐéð¥ ðÀð┤ðÁÐüÐî Ðüð¥ð┤ðÁÐÇðÂð©ÐéÐüÐÅ ð©ð¢Ðäð¥ÐÇð╝ð░Ðåð©ÐÅ, ð┐ÐÇðÁð┤ð¢ð░ðÀð¢ð░ÐçðÁð¢ð¢ð░ÐÅ ð©Ðüð║ð╗ÐÄÐçð©ÐéðÁð╗Ðîð¢ð¥ ð┤ð╗ÐÅ Ðüð┐ðÁÐåð©ð░ð╗ð©ÐüÐéð¥ð▓ ðÀð┤ÐÇð░ð▓ð¥ð¥ÐàÐÇð░ð¢ðÁð¢ð©ÐÅ.
ðòÐüð╗ð© ðÆÐï ð¢ðÁ ÐÅð▓ð╗ÐÅðÁÐéðÁÐüÐî Ðüð┐ðÁÐåð©ð░ð╗ð©ÐüÐéð¥ð╝ ðÀð┤ÐÇð░ð▓ð¥ð¥ÐàÐÇð░ð¢ðÁð¢ð©ÐÅ, ð░ð┤ð╝ð©ð¢ð©ÐüÐéÐÇð░Ðåð©ÐÅ ð¢ðÁ ð¢ðÁÐüðÁÐé ð¥Ðéð▓ðÁÐéÐüÐéð▓ðÁð¢ð¢ð¥ÐüÐéð© ðÀð░ ð▓ð¥ðÀð╝ð¥ðÂð¢ÐïðÁ ð¥ÐéÐÇð©Ðåð░ÐéðÁð╗Ðîð¢ÐïðÁ ð┐ð¥Ðüð╗ðÁð┤ÐüÐéð▓ð©ÐÅ, ð▓ð¥ðÀð¢ð©ð║Ðêð©ðÁ ð▓ ÐÇðÁðÀÐâð╗ÐîÐéð░ÐéðÁ Ðüð░ð╝ð¥ÐüÐéð¥ÐÅÐéðÁð╗Ðîð¢ð¥ð│ð¥ ð©Ðüð┐ð¥ð╗ÐîðÀð¥ð▓ð░ð¢ð©ÐÅ ðÆð░ð╝ð© ð©ð¢Ðäð¥ÐÇð╝ð░Ðåð©ð© Ðü ð┐ð¥ÐÇÐéð░ð╗ð░ ð▒ðÁðÀ ð┐ÐÇðÁð┤ð▓ð░ÐÇð©ÐéðÁð╗Ðîð¢ð¥ð╣ ð║ð¥ð¢ÐüÐâð╗ÐîÐéð░Ðåð©ð© Ðü ð▓ÐÇð░Ðçð¥ð╝.
ðØð░ðÂð©ð╝ð░ÐÅ ð¢ð░ ð║ð¢ð¥ð┐ð║Ðâ ┬½ðÆð¥ð╣Ðéð©┬╗, ðÆÐï ð┐ð¥ð┤Ðéð▓ðÁÐÇðÂð┤ð░ðÁÐéðÁ, ÐçÐéð¥ ÐÅð▓ð╗ÐÅðÁÐéðÁÐüÐî ð▓ÐÇð░Ðçð¥ð╝ ð©ð╗ð© ÐüÐéÐâð┤ðÁð¢Ðéð¥ð╝ ð╝ðÁð┤ð©Ðåð©ð¢Ðüð║ð¥ð│ð¥ ð▓ÐâðÀð░.