–Ф–µ–Љ–µ–љ—Ж–Є—П –њ—А–Є –±–Њ–ї–µ–Ј–љ–Є –Я–∞—А–Ї–Є–љ—Б–Њ–љ–∞
- –Р–љ–љ–Њ—В–∞—Ж–Є—П
- –°—В–∞—В—М—П
- –°—Б—Л–ї–Ї–Є
- English

–Ю–њ—А–µ–і–µ–ї–µ–љ–Є–µ. –≠–њ–Є–і–µ–Љ–Є–Њ–ї–Њ–≥–Є—П. –Ю—Б–Њ–±–µ–љ–љ–Њ—Б—В–Є —В–µ—З–µ–љ–Є—П
–С–Њ–ї–µ–Ј–љ—М –Я–∞—А–Ї–Є–љ—Б–Њ–љ–∞ (–С–Я)¬†вАУ –Љ—Г–ї—М—В–Є—Б–Є—Б—В–µ–Љ–љ–Њ–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ, –њ—А–Њ—П–≤–ї—П—О—Й–µ–µ—Б—П –Љ–Њ—В–Њ—А–љ—Л–Љ–Є –Є¬†–Љ–љ–Њ–≥–Њ—З–Є—Б–ї–µ–љ–љ—Л–Љ–Є –љ–µ–Љ–Њ—В–Њ—А–љ—Л–Љ–Є —Б–Є–Љ–њ—В–Њ–Љ–∞–Љ–Є, –≤–∞–ґ–љ–Њ–µ –Љ–µ—Б—В–Њ —Б—А–µ–і–Є –Ї–Њ—В–Њ—А—Л—Е –Ј–∞–љ–Є–Љ–∞—О—В –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л–µ —А–∞—Б—Б—В—А–Њ–є—Б—В–≤–∞.
–°–Њ–≥–ї–∞—Б–љ–Њ –Ф–Є–∞–≥–љ–Њ—Б—В–Є—З–µ—Б–Ї–Њ–Љ—Г –Є¬†—Б—В–∞—В–Є—Б—В–Є—З–µ—Б–Ї–Њ–Љ—Г —А—Г–Ї–Њ–≤–Њ–і—Б—В–≤—Г –њ–Њ¬†–њ—Б–Є—Е–Є—З–µ—Б–Ї–Є–Љ —А–∞—Б—Б—В—А–Њ–є—Б—В–≤–∞–Љ (Diagnostic and Statistical Manual of¬†Mental Disorders IV¬†вАУ DSM-IV), –і–µ–Љ–µ–љ—Ж–Є—П¬†вАУ –њ—А–Є–Њ–±—А–µ—В–µ–љ–љ–Њ–µ —Б–љ–Є–ґ–µ–љ–Є–µ –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л—Е —Д—Г–љ–Ї—Ж–Є–є, –Ї–Њ—В–Њ—А–Њ–µ –≤—Л–Ј–≤–∞–љ–Њ –Њ—А–≥–∞–љ–Є—З–µ—Б–Ї–Є–Љ –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є–µ–Љ¬†–≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞, –Њ–њ—А–µ–і–µ–ї—П–µ—В—Б—П –љ–∞¬†—Д–Њ–љ–µ —П—Б–љ–Њ–≥–Њ —Б–Њ–Ј–љ–∞–љ–Є—П –Є¬†–њ—А–Є–≤–Њ–і–Є—В –Ї¬†–Њ–≥—А–∞–љ–Є—З–µ–љ–Є—О –њ–Њ–≤—Б–µ–і–љ–µ–≤–љ–Њ–є –ґ–Є–Ј–љ–µ–і–µ—П—В–µ–ї—М–љ–Њ—Б—В–Є.
–Я–Њ¬†—А–µ–Ј—Г–ї—М—В–∞—В–∞–Љ –њ—А–Њ–≤–µ–і–µ–љ–љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є, –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л–µ –љ–∞—А—Г—И–µ–љ–Є—П —А–∞–Ј–љ–Њ–є —Б—В–µ–њ–µ–љ–Є –≤—Л—А–∞–ґ–µ–љ–љ–Њ—Б—В–Є (–ї–µ–≥–Ї–Є–µ, —Г–Љ–µ—А–µ–љ–љ—Л–µ, —В—П–ґ–µ–ї—Л–µ) –Њ–±–љ–∞—А—Г–ґ–Є–≤–∞—О—В—Б—П –њ—А–Є –љ–µ–є—А–Њ–њ—Б–Є—Е–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–Љ –Њ–±—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є —Г¬†90вАУ95% –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–С–Я [1]. –Ь–µ—В–∞–∞–љ–∞–ї–Є–Ј 13 –њ–Њ–њ—Г–ї—П—Ж–Є–Њ–љ–љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –њ–Њ–Ї–∞–Ј–∞–ї, —З—В–Њ —Б—А–µ–і–љ—П—П —А–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–љ–Њ—Б—В—М –і–µ–Љ–µ–љ—Ж–Є–Є —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–С–Я —А–∞–≤–љ–∞ 31,5% [2]. –°–Њ–≥–ї–∞—Б–љ–Њ –і–∞–љ–љ—Л–Љ –ї–Њ–љ–≥–Є—В—Г–і–Є–љ–∞–ї—М–љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є, –Ї—Г–Љ—Г–ї—П—В–Є–≤–љ–∞—П —А–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–љ–Њ—Б—В—М –і–µ–Љ–µ–љ—Ж–Є–Є —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–С–Я —Б–Њ—Б—В–∞–≤–ї—П–µ—В 90% [3], —З—В–Њ –і–µ–ї–∞–µ—В —Б–≤—П–Ј—М –і–µ–Љ–µ–љ—Ж–Є–Є —Б¬†–С–Я –њ–Њ—З—В–Є –љ–µ–Є–Ј–±–µ–ґ–љ–Њ–є.
–Ъ–∞–Ї —Г—Б—В–∞–љ–Њ–≤–Є–ї–Є T.A. Hughes (2000) –Є¬†D. Aarsland (2003), –Њ—В¬†–Љ–Њ–Љ–µ–љ—В–∞ —А–∞–Ј–≤–Є—В–Є—П –Љ–Њ—В–Њ—А–љ—Л—Е –њ—А–Њ—П–≤–ї–µ–љ–Є–є –С–Я –і–Њ —А–∞–Ј–≤–Є—В–Є—П –і–µ–Љ–µ–љ—Ж–Є–Є –≤¬†—Б—А–µ–і–љ–µ–Љ –њ—А–Њ—Е–Њ–і–Є—В –і–µ—Б—П—В—М –ї–µ—В [4], –љ–Њ¬†—Н—В–Њ—В –≤—А–µ–Љ–µ–љ–љ–Њ–є –Є–љ—В–µ—А–≤–∞–ї —И–Є—А–Њ–Ї–Њ –≤–∞—А—М–Є—А—Г–µ—В—Б—П. –£¬†–љ–µ–Ї–Њ—В–Њ—А—Л—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –і–µ–Љ–µ–љ—Ж–Є—П —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П –≤—Б–Ї–Њ—А–µ –њ–Њ—Б–ї–µ –і–µ–±—О—В–∞ –њ–∞—А–Ї–Є–љ—Б–Њ–љ–Є–Ј–Љ–∞, —Г¬†–і—А—Г–≥–Є—Е, –љ–∞–њ—А–Њ—В–Є–≤, –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л–µ –љ–∞—А—Г—И–µ–љ–Є—П, –і–Њ—Б—В–Є–≥–∞—О—Й–Є–µ —Б—В–µ–њ–µ–љ–Є –і–µ–Љ–µ–љ—Ж–Є–Є, –≤–Њ–Ј–љ–Є–Ї–∞—О—В —З–µ—А–µ–Ј 20 –Є¬†–±–Њ–ї–µ–µ –ї–µ—В [5]. –Р. Schrag –Є¬†—Б–Њ–∞–≤—В. (1998) —Б—З–Є—В–∞—О—В, —З—В–Њ –≤–Њ–Ј—А–∞—Б—В¬†вАУ –Ї–ї—О—З–µ–≤–Њ–є —Д–∞–Ї—В–Њ—А —А–∞–Ј–≤–Є—В–Є—П –і–µ–Љ–µ–љ—Ж–Є–Є. –£¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†—А–∞–љ–љ–Є–Љ –љ–∞—З–∞–ї–Њ–Љ –С–Я –і–µ–Љ–µ–љ—Ж–Є—П —А–µ–і–Ї–Њ —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П –≤¬†—В–µ—З–µ–љ–Є–µ –њ–µ—А–≤—Л—Е –і–µ—Б—П—В–Є –ї–µ—В –±–Њ–ї–µ–Ј–љ–Є [6].
–†–∞–Ј–≤–Є–≤—И–Є—Б—М, –і–µ–Љ–µ–љ—Ж–Є—П –њ—А–Є –С–Я –Њ–±—Л—З–љ–Њ –љ–µ—Г–Ї–ї–Њ–љ–љ–Њ –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г–µ—В. D. Aarsland (2004) —Г—Б—В–∞–љ–Њ–≤–Є–ї, —З—В–Њ –µ–ґ–µ–≥–Њ–і–љ–Њ–µ —Г—Е—Г–і—И–µ–љ–Є–µ —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–і–µ–Љ–µ–љ—Ж–Є–µ–є –њ—А–Є –С–Я —Б–Њ—Б—В–∞–≤–ї—П–µ—В –≤¬†—Б—А–µ–і–љ–µ–Љ 2,3 –±–∞–ї–ї–∞ –њ–Њ¬†–Ъ—А–∞—В–Ї–Њ–є —И–Ї–∞–ї–µ –Њ—Ж–µ–љ–Ї–Є –њ—Б–Є—Е–Є—З–µ—Б–Ї–Њ–≥–Њ —Б—В–∞—В—Г—Б–∞ (–і–Є–љ–∞–Љ–Є–Ї–∞ —Б–Њ—Б—В–Њ—П–љ–Є—П –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –Њ—Ж–µ–љ–Є–≤–∞–ї–∞—Б—М –≤¬†—В–µ—З–µ–љ–Є–µ —З–µ—В—Л—А–µ—Е –ї–µ—В). –Т¬†—Ж–µ–ї–Њ–Љ —Б–Ї–Њ—А–Њ—Б—В—М –њ—А–Њ–≥—А–µ—Б—Б–Є–Є —Б–Њ–њ–Њ—Б—В–∞–≤–Є–Љ–∞ —Б¬†—А–∞–Ј–≤–Є—В–Є–µ–Љ –і–µ–Љ–µ–љ—Ж–Є–Є –њ—А–Є –і–µ–Љ–µ–љ—Ж–Є–Є —Б¬†—В–µ–ї—М—Ж–∞–Љ–Є –Ы–µ–≤–Є [7] –Є¬†–Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ –њ—А–µ–≤–Њ—Б—Е–Њ–і–Є—В —В–∞–Ї–Њ–≤—Г—О —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–С–Я –±–µ–Ј –і–µ–Љ–µ–љ—Ж–Є–Є (–Њ–Ї–Њ–ї–Њ 1 –±–∞–ї–ї–∞ –њ–Њ¬†–Ъ—А–∞—В–Ї–Њ–є —И–Ї–∞–ї–µ –Њ—Ж–µ–љ–Ї–Є –њ—Б–Є—Е–Є—З–µ—Б–Ї–Њ–≥–Њ —Б—В–∞—В—Г—Б–∞ –≤¬†–≥–Њ–і) [5].
–§–∞–Ї—В–Њ—А—Л —А–Є—Б–Ї–∞
–Ъ¬†—Д–∞–Ї—В–Њ—А–∞–Љ —А–Є—Б–Ї–∞ —А–∞–Ј–≤–Є—В–Є—П –і–µ–Љ–µ–љ—Ж–Є–Є –њ—А–Є –С–Я –Њ—В–љ–Њ—Б—П—В:
- –њ–Њ–ґ–Є–ї–Њ–є –≤–Њ–Ј—А–∞—Б—В [8];
- —В—П–ґ–µ–ї—Л–є –њ–∞—А–Ї–Є–љ—Б–Њ–љ–Є–Ј–Љ, –Њ—Б–Њ–±–µ–љ–љ–Њ —А–Є–≥–Є–і–љ–Њ—Б—В—М, –њ–Њ—Б—В—Г—А–∞–ї—М–љ—Г—О –љ–µ—Г—Б—В–Њ–є—З–Є–≤–Њ—Б—В—М –Є¬†–љ–∞—А—Г—И–µ–љ–Є—П –њ–Њ—Е–Њ–і–Ї–Є [8вАУ10], —В—А–∞–љ—Б—Д–Њ—А–Љ–∞—Ж–Є—О –і—А–Њ–ґ–∞—В–µ–ї—М–љ–Њ–є —Д–Њ—А–Љ—Л –С–Я –≤¬†–∞–Ї–Є–љ–µ—В–Є–Ї–Њ-—А–Є–≥–Є–і–љ—Г—О [7];
- –ї–µ–≥–Ї–Њ–µ –Ї–Њ–≥–љ–Є—В–Є–≤–љ–Њ–µ —Б–љ–Є–ґ–µ–љ–Є–µ –≤¬†–і–µ–±—О—В–µ –С–Я [10] —Б¬†–і–µ—Д–Є—Ж–Є—В–Њ–Љ –Ї–Њ—А—В–Є–Ї–∞–ї—М–љ—Л—Е —Д—Г–љ–Ї—Ж–Є–є –≤¬†–Ї–Њ–≥–љ–Є—В–Є–≤–љ–Њ–Љ –њ—А–Њ—Д–Є–ї–µ, –ї–Њ–Ї–∞–ї–Є–Ј—Г—О—Й–Є—Е—Б—П –≤¬†–≤–Є—Б–Њ—З–љ–Њ-—В–µ–Љ–µ–љ–љ–Њ-–Ј–∞—В—Л–ї–Њ—З–љ—Л—Е –Њ—В–і–µ–ї–∞—Е –Ї–Њ—А—Л¬†–≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞: —Б–µ–Љ–∞–љ—В–Є—З–µ—Б–Ї–∞—П –∞—Д–∞–Ј–Є—П, –Ј—А–Є—В–µ–ї—М–љ–Њ-–њ—А–Њ—Б—В—А–∞–љ—Б—В–≤–µ–љ–љ—Л–µ –љ–∞—А—Г—И–µ–љ–Є—П. –Я—А–Є —Н—В–Њ–Љ –і–µ—Д–Є—Ж–Є—В –Є—Б–њ–Њ–ї–љ–Є—В–µ–ї—М–љ—Л—Е —Д—Г–љ–Ї—Ж–Є–є –љ–µ¬†–∞—Б—Б–Њ—Ж–Є–Є—А—Г–µ—В—Б—П —Б¬†–≤—Л—Б–Њ–Ї–Є–Љ —А–Є—Б–Ї–Њ–Љ —А–∞–Ј–≤–Є—В–Є—П –і–µ–Љ–µ–љ—Ж–Є–Є. –Ю–њ–Є—Б–∞–љ–љ—Л–µ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ –і–∞–љ–љ—Л–µ –њ–Њ–і–Ї—А–µ–њ–ї–µ–љ—Л —Б–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Є–Љ–Є¬†–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є–Љ–Є –Є–Ј–Љ–µ–љ–µ–љ–Є—П–Љ–Є, –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–љ—Л–Љ–Є –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–Љ –њ–∞—В—В–µ—А–љ–Њ–Љ. –Ґ–∞–Ї, –≥–∞–њ–ї–Њ—В–Є–њ H1 –≤ –≥–µ–љ–µ —В–∞—Г-–њ—А–Њ—В–µ–Є–љ–∞ —Б–≤—П–Ј–∞–љ —Б¬†–Ј–∞–і–љ–Є–Љ –Ї–Њ—А–Ї–Њ–≤—Л–Љ –і–µ—Д–Є—Ж–Є—В–Њ–Љ –Є¬†–≤—Л—Б–Њ–Ї–Є–Љ —А–Є—Б–Ї–Њ–Љ —А–∞–Ј–≤–Є—В–Є—П –і–µ–Љ–µ–љ—Ж–Є–Є, –≤¬†—В–Њ –≤—А–µ–Љ—П –Ї–∞–Ї¬†–≥–µ–љ–Њ—В–Є–њ –Ї–∞—В–µ—Е–Њ–ї-–Ю-–Љ–µ—В–Є–ї—В—А–∞–љ—Б—Д–µ—А–∞–Ј—Л –∞—Б—Б–Њ—Ж–Є–Є—А–Њ–≤–∞–ї—Б—П —Б¬†–љ–∞—А—Г—И–µ–љ–Є–µ–Љ –Є—Б–њ–Њ–ї–љ–Є—В–µ–ї—М–љ—Л—Е —Д—Г–љ–Ї—Ж–Є–є –Є¬†–љ–µ —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞–ї—Б—П —А–Є—Б–Ї–Њ–Љ —А–∞–Ј–≤–Є—В–Є—П –і–µ–Љ–µ–љ—Ж–Є–Є [10];
- —А–∞–љ–љ–µ–µ —А–∞–Ј–≤–Є—В–Є–µ –Ј—А–Є—В–µ–ї—М–љ—Л—Е¬†–≥–∞–ї–ї—О—Ж–Є–љ–∞—Ж–Є–є –Є¬†–Є–ї–ї—О–Ј–Є–є, —З—В–Њ, –≤–µ—А–Њ—П—В–љ–Њ, —Б–≤—П–Ј–∞–љ–Њ —Б¬†–Њ—В–ї–Њ–ґ–µ–љ–Є–µ–Љ —В–µ–ї–µ—Ж –Ы–µ–≤–Є –≤¬†–≤–Є—Б–Њ—З–љ–Њ–є –Ї–Њ—А–µ, –Љ–Є–љ–і–∞–ї–Є–љ–µ –Є¬†—А–∞–Ј–≤–Є—В–Є–µ–Љ —Е–Њ–ї–Є–љ–µ—А–≥–Є—З–µ—Б–Ї–Њ–≥–Њ –і–µ—Д–Є—Ж–Є—В–∞ [11,¬†12];
- –∞–њ–∞—В–Є—О [8, 13];
- –љ–∞—А—Г—И–µ–љ–Є–µ –њ–Њ–≤–µ–і–µ–љ–Є—П –≤¬†—Д–∞–Ј—Г –±—Л—Б—В—А–Њ–≥–Њ —Б–љ–∞ (–Њ—В—Б—Г—В—Б—В–≤–Є–µ –∞—В–Њ–љ–Є–Є –≤¬†—Д–∞–Ј—Г –±—Л—Б—В—А–Њ–≥–Њ —Б–љ–∞) [14];
- –і–ї–Є—В–µ–ї—М–љ–Њ–µ –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є–µ —Е–Њ–ї–Є–љ–Њ–ї–Є—В–Є–Ї–Њ–≤ (—Ж–Є–Ї–ї–Њ–і–Њ–ї–∞, –∞–Ї–Є–љ–µ—В–Њ–љ–∞), –≤¬†—В–Њ–Љ —З–Є—Б–ї–µ –∞–љ—В–Є–і–µ–њ—А–µ—Б—Б–∞–љ—В–Њ–≤ —Б¬†—Е–Њ–ї–Є–љ–Њ–ї–Є—В–Є—З–µ—Б–Ї–Њ–є –∞–Ї—В–Є–≤–љ–Њ—Б—В—М—О (–∞–Љ–Є—В—А–Є–њ—В–Є–ї–Є–љ–∞) [15], –≤¬†—Б–≤—П–Ј–Є —Б¬†—Г—Б–Ї–Њ—А–µ–љ–Є–µ–Љ –∞–Љ–Є–ї–Њ–Є–і–Њ–≥–µ–љ–µ–Ј–∞ –љ–∞¬†—Д–Њ–љ–µ –Є—Е –њ—А–Є–µ–Љ–∞ [12];
- —Б–µ–Љ–µ–є–љ—Л–є –∞–љ–∞–Љ–љ–µ–Ј (—Б—В—А–∞–і–∞—О—Й–Є–µ –і–µ–Љ–µ–љ—Ж–Є–µ–є –±–ї–Є–Ј–Ї–Є–µ —А–Њ–і—Б—В–≤–µ–љ–љ–Є–Ї–Є) [4].
–У–µ–љ–µ—В–Є—З–µ—Б–Ї–Є–µ —Д–∞–Ї—В–Њ—А—Л —А–Є—Б–Ї–∞
–Ю—В–Ї—А—Л—В–Є–µ –ї–Њ–Ї—Г—Б–Њ–≤¬†–≥–µ–љ–Њ–≤, –Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ—Л—Е –Ј–∞ —А–∞–Ј–≤–Є—В–Є–µ –љ–∞—Б–ї–µ–і—Б—В–≤–µ–љ–љ—Л—Е —Д–Њ—А–Љ –С–Я, –њ–Њ–Ј–≤–Њ–ї–Є–ї–Њ –њ–Њ-–љ–Њ–≤–Њ–Љ—Г –≤–Ј–≥–ї—П–љ—Г—В—М –љ–∞¬†–њ—А–Њ–±–ї–µ–Љ—Л —Н—В–Є–Њ–њ–∞—В–Њ–≥–µ–љ–µ–Ј–∞ –Є¬†–і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–Є –С–Я. –•–Њ—В—П, –Ї–∞–Ї –њ—А–∞–≤–Є–ї–Њ, –С–Я –љ–Њ—Б–Є—В —Б–њ–Њ—А–∞–і–Є—З–µ—Б–Ї–Є–є —Е–∞—А–∞–Ї—В–µ—А, —Г¬†15% –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –і–Є–∞–≥–љ–Њ—Б—В–Є—А—Г–µ—В—Б—П —Б–µ–Љ–µ–є–љ–∞—П —Д–Њ—А–Љ–∞ –С–Я, –Ї–Њ–≥–і–∞ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ –Є–Љ–µ–µ—В –Љ–µ—Б—В–Њ —Г¬†–љ–µ—Б–Ї–Њ–ї—М–Ї–Є—Е —А–Њ–і—Б—В–≤–µ–љ–љ–Є–Ї–Њ–≤ –Є–Ј¬†–Њ–і–љ–Њ–≥–Њ –Є–ї–Є —А–∞–Ј–љ—Л—Е –њ–Њ–Ї–Њ–ї–µ–љ–Є–є. –Ф–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ –Љ—Г—В–∞—Ж–Є–Є –њ–Њ¬†–Ї—А–∞–є–љ–µ–є –Љ–µ—А–µ –≤¬†–њ—П—В–Є¬†–≥–µ–љ–∞—Е –Њ–њ—А–µ–і–µ–ї–µ–љ–љ–Њ –њ—А–Є–≤–Њ–і—П—В –Ї¬†—А–∞–Ј–≤–Є—В–Є—О –Љ–µ–љ–і–µ–ї–Є—А—Г—О—Й–Є—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є [16].
–У–µ–љ—Л –∞–ї—М—Д–∞-—Б–Є–љ—Г–Ї–ї–µ–Є–љ–∞ (SNCA) –Є¬†–Њ–±–Њ–≥–∞—Й–µ–љ–љ–Њ–є –ї–µ–є—Ж–Є–љ–Њ–≤—Л–Љ–Є –њ–Њ–≤—В–Њ—А–∞–Љ–Є –Ї–Є–љ–∞–Ј—Л 2 (Leucine-Rich Repeat Kinase 2¬†вАУ LRRK2) –∞—Б—Б–Њ—Ж–Є–Є—А–Њ–≤–∞–љ—Л —Б¬†—А–∞–Ј–≤–Є—В–Є–µ–Љ –∞—Г—В–Њ—Б–Њ–Љ–љ–Њ-–і–Њ–Љ–Є–љ–∞–љ—В–љ—Л—Е —Д–Њ—А–Љ –С–Я [16].
–Ь–Є—Б—Б–µ–љ—Б-–Љ—Г—В–∞—Ж–Є–Є¬†–≥–µ–љ–∞ SNCA —Б–≤—П–Ј–∞–љ—Л —Б¬†–±—Л—Б—В—А—Л–Љ –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ–Љ –С–Я, –∞¬†—В–∞–Ї–ґ–µ —Б¬†–≤—Л—Б–Њ–Ї–Њ–є —А–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–љ–Њ—Б—В—М—О –і–µ–Љ–µ–љ—Ж–Є–Є –Є¬†–і—А—Г–≥–Є—Е –њ—Б–Є—Е–Њ—В–Є—З–µ—Б–Ї–Є—Е —Б–Є–Љ–њ—В–Њ–Љ–Њ–≤. –Ь–љ–Њ–ґ–µ—Б—В–≤–µ–љ–љ—Л–µ –њ–Њ–≤—В–Њ—А—Л SNCA –≤—Л—П–≤–ї—П—О—В—Б—П –Є¬†–њ—А–Є —Б–µ–Љ–µ–є–љ—Л—Е —Д–Њ—А–Љ–∞—Е –С–Я, –Є¬†–њ—А–Є —Б–њ–Њ—А–∞–і–Є—З–µ—Б–Ї–Є—Е —Б–ї—Г—З–∞—П—Е –С–Я. –Ъ–ї–Є–љ–Є—З–µ—Б–Ї–∞—П –Ї–∞—А—В–Є–љ–∞ –С–Я –Ј–∞–≤–Є—Б–Є—В –Њ—В¬†–Ї–Њ–ї–Є—З–µ—Б—В–≤–∞ –њ–Њ–≤—В–Њ—А–Њ–≤ SNCA. –Э–∞–њ—А–Є–Љ–µ—А, –њ–∞—Ж–Є–µ–љ—В—Л —Б¬†—В—А–Є–њ–ї–Є–Ї–∞—Ж–Є–µ–є SNCA –і–µ–Љ–Њ–љ—Б—В—А–Є—А—Г—О—В —А–∞–љ–љ–µ–µ –љ–∞—З–∞–ї–Њ –±–Њ–ї–µ–Ј–љ–Є –Є¬†–≤—Л—Б–Њ–Ї—Г—О —Б–Ї–Њ—А–Њ—Б—В—М —А–∞–Ј–≤–Є—В–Є—П –і–µ–Љ–µ–љ—Ж–Є–Є, –≤¬†—В–Њ –≤—А–µ–Љ—П –Ї–∞–Ї —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–і—Г–њ–ї–Є–Ї–∞—Ж–Є–µ–є SNCA –С–Я –љ–∞—З–Є–љ–∞–µ—В—Б—П –њ–Њ–Ј–і–љ–µ–µ –Є¬†—А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П —В–Є–њ–Є—З–љ–Њ.
–Я–∞—Ж–Є–µ–љ—В—Л —Б¬†–С–Я¬†вАУ –љ–Њ—Б–Є—В–µ–ї–Є –Љ—Г—В–∞—Ж–Є–Є LRRK2 –Є–Љ–µ—О—В –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–є –њ–∞—В—В–µ—А–љ, –њ–Њ—Е–Њ–ґ–Є–є –љ–∞¬†—Б–њ–Њ—А–∞–і–Є—З–µ—Б–Ї—Г—О –С–Я, –љ–Њ¬†—Б –љ–µ—Б–Ї–Њ–ї—М–Ї–Њ –±–Њ–ї–µ–µ –і–Њ–±—А–Њ–Ї–∞—З–µ—Б—В–≤–µ–љ–љ–Њ–є –њ—А–Њ–≥—А–µ—Б—Б–Є–µ–є –Є¬†–Љ–µ–і–ї–µ–љ–љ—Л–Љ —А–∞–Ј–≤–Є—В–Є–µ–Љ –і–µ¬≠–Љ–µ–љ—Ж–Є–Є.
–У–µ–љ—Л –њ–∞—А–Ї–Є–љ–∞ (PARK2), DJ-1, PINK1 –∞—Б—Б–Њ—Ж–Є–Є—А–Њ–≤–∞–љ—Л c –∞—Г—В–Њ—Б–Њ–Љ–љ–Њ-—А–µ—Ж–µ—Б—Б–Є–≤–љ–Њ–є —Д–Њ—А–Љ–Њ–є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П —Б¬†—А–∞–љ–љ–Є–Љ –љ–∞—З–∞–ї–Њ–Љ [16, 17]. –Ф–ї—П PINK1 –Є¬†DJ-1 —В–Є–њ–Є—З–љ—Л —А–∞–љ–љ–µ–µ –љ–∞—З–∞–ї–Њ, –Љ–µ–і–ї–µ–љ–љ–Њ–µ –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–µ –С–Я –Є¬†–Љ–µ–і–ї–µ–љ–љ–Њ–µ —А–∞–Ј–≤–Є—В–Є–µ –і–µ–Љ–µ–љ—Ж–Є–Є.
–Ъ¬†–≥–µ–љ–∞–Љ –≤—Л—Б–Њ–Ї–Њ–≥–Њ —А–Є—Б–Ї–∞ –С–Я –Њ—В–љ–Њ—Б—П—В¬†–≥–µ–љ¬†–≥–ї—О–Ї–Њ—Ж–µ—А–µ–±—А–Њ–Ј–Є–і–∞–Ј—Л [18], –Љ—Г—В–∞—Ж–Є–Є –Ї–Њ—В–Њ—А–Њ–≥–Њ –≤¬†2,4 —А–∞–Ј–∞ —Г–≤–µ–ї–Є—З–Є–≤–∞—О—В —А–Є—Б–Ї¬†–Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л—Е –љ–∞—А—Г—И–µ–љ–Є–є [19].
–Т¬†—В–Њ –ґ–µ –≤—А–µ–Љ—П –њ–Њ–ї–Є–Љ–Њ—А—Д–Є–Ј–Љ—Л —Б–≤—П–Ј–∞–љ–љ–Њ–≥–Њ —Б¬†–Љ–Є–Ї—А–Њ—В—А—Г–±–Њ—З–Ї–∞–Љ–Є —В–∞—Г-–њ—А–Њ—В–µ–Є–љ–∞ (Microtubule-Associated Protein Tau), –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ, –Є–Љ–µ—О—В –љ–µ–Ї–Њ—В–Њ—А–Њ–µ –Ј–љ–∞—З–µ–љ–Є–µ –і–ї—П —А–∞–Ј–≤–Є—В–Є—П –і–µ–Љ–µ–љ—Ж–Є–Є –њ—А–Є –С–Я [19].
–Ч–∞—Й–Є—В–љ—Л–µ —Д–∞–Ї—В–Њ—А—Л
–Я—А–µ–і–њ–Њ–ї–∞–≥–∞–µ—В—Б—П, —З—В–Њ –љ–µ–Ї–Њ—В–Њ—А—Л–µ —Д–∞–Ї—В–Њ—А—Л –Љ–Њ–≥—Г—В –Ј–∞—Й–Є—В–Є—В—М –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б –±–Њ–ї–µ–Ј–љ—М—О –Я–∞—А–Ї–Є–љ—Б–Њ–љ–∞ –Њ—В —А–∞–Ј–≤–Є—В–Є—П –і–µ–Љ–µ–љ—Ж–Є–Є. –Ю—Б–Њ–±—Л–є –Є–љ—В–µ—А–µ—Б –њ—А–µ–і—Б—В–∞–≤–ї—П—О—В –і–∞–љ–љ—Л–µ –Њ¬†–Ј–∞—Й–Є—В–љ–Њ–Љ –і–µ–є—Б—В–≤–Є–Є –љ–Њ—А–Љ–∞–ї—М–љ–Њ–≥–Њ –Ї–Є—И–µ—З–љ–Њ–≥–Њ –Љ–Є–Ї—А–Њ–±–Є–Њ–Љ–∞, –≤–µ–і—Г—Й–µ–≥–Њ –Ї¬†—Б–љ–Є–ґ–µ–љ–Є—О –њ—А–Њ–љ–Є—Ж–∞–µ–Љ–Њ—Б—В–Є –Є–љ—В–µ—А—Б—В–Є—Ж–Є–∞–ї—М–љ–Њ–≥–Њ —Н–њ–Є—В–µ–ї–Є–∞–ї—М–љ–Њ–≥–Њ –±–∞—А—М–µ—А–∞ –Є¬†–Ї–Њ–љ—В—А–Њ–ї—О –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Є—Е –Є–Љ–Љ—Г–љ–љ—Л—Е —А–µ–∞–Ї—Ж–Є–є, –≤¬†–њ—А–Њ—В–Є–≤–Њ–≤–µ—Б —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–Љ—Г –≤–Њ—Б–њ–∞–ї–µ–љ–Є—О –≤¬†–ґ–µ–ї—Г–і–Њ—З–љ–Њ-–Ї–Є—И–µ—З–љ–Њ–Љ —В—А–∞–Ї—В–µ, –Ї–Њ—В–Њ—А–Њ–µ, –њ–Њ¬†–Љ–љ–µ–љ–Є—О T.R.¬†Sampson (2016), —П–≤–ї—П–µ—В—Б—П —В—А–Є–≥–≥–µ—А–Њ–Љ –љ–µ–є—А–Њ–і–µ–≥–µ–љ–µ—А–∞—В–Є–≤–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є, –≤¬†—В–Њ–Љ —З–Є—Б–ї–µ –С–Я [20].
–Х—Б—В—М –Љ–љ–µ–љ–Є–µ, —З—В–Њ —Б–љ–Є–ґ–µ–љ–Є–µ –Є–љ—Д–µ–Ї—Ж–Є–Њ–љ–љ–Њ–є –љ–∞–≥—А—Г–Ј–Ї–Є –љ–∞¬†–Њ—А–≥–∞–љ–Є–Ј–Љ –Ј–∞ —Б—З–µ—В —Н–ї–Є–Љ–Є–љ–∞—Ж–Є–Є –±–∞–Ї—В–µ—А–Є–є –Є¬†–њ–Њ–і–∞–≤–ї–µ–љ–Є—П —А–µ–њ–ї–Є–Ї–∞—Ж–Є–Є –≤–Є—А—Г—Б–Њ–≤, –≤—Л–Ј—Л–≤–∞—О—Й–Є—Е —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–µ –≤–Њ—Б–њ–∞–ї–µ–љ–Є–µ, –Њ–±–µ—Б–њ–µ—З–Є–≤–∞–µ—В —Г–Љ–µ–љ—М—И–µ–љ–Є–µ —А–Є—Б–Ї–∞ –і–µ–≥–µ–љ–µ—А–∞—В–Є–≤–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є –љ–µ—А–≤–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л [21].
–Т¬†—Е–Њ–і–µ –ї–Њ–љ–≥–Є—В—Г–і–Є–љ–∞–ї—М–љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –±—Л–ї–Є –њ–Њ–ї—Г—З–µ–љ—Л –њ—А–Њ—В–Є–≤–Њ—А–µ—З–Є–≤—Л–µ —А–µ–Ј—Г–ї—М—В–∞—В—Л –Њ¬†–њ—А–Њ—В–µ–Ї—В–Є–≤–љ–Њ–Љ –і–µ–є—Б—В–≤–Є–Є –Ї—Г—А–µ–љ–Є—П –≤¬†–Њ—В–љ–Њ—И–µ–љ–Є–Є —А–∞–Ј–≤–Є—В–Є—П –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л—Е –љ–∞—А—Г—И–µ–љ–Є–є –њ—А–Є –С–Я [22]. –Ґ–∞–Ї, –≤–Њ–Ј–і–µ–є—Б—В–≤–Є–µ –љ–∞¬†–љ–Є–Ї–Њ—В–Є–љ–Њ–≤—Л–µ —А–µ—Ж–µ–њ—В–Њ—А—Л, —Г—З–∞—Б—В–≤—Г—О—Й–Є–µ –≤¬†–Њ–±—Г—З–µ–љ–Є–Є, –Љ–Њ–ґ–µ—В –Ј–∞–Љ–µ–і–ї—П—В—М –Ї–Њ–≥–љ–Є—В–Є–≤–љ–Њ–µ —Б–љ–Є–ґ–µ–љ–Є–µ, –Њ–і–љ–∞–Ї–Њ –Ї—Г—А–µ–љ–Є–µ —Г—Б–Є–ї–Є–≤–∞–µ—В –Њ–Ї–Є—Б–ї–Є—В–µ–ї—М–љ—Л–є —Б—В—А–µ—Б—Б –Є¬†–њ—А–Њ–≤–Њ—Ж–Є—А—Г–µ—В —А–∞–Ј–≤–Є—В–Є–µ —Б–µ—А–і–µ—З–љ–Њ-—Б–Њ—Б—Г–і–Є—Б—В—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є.
R. Inzelberg –Є¬†—Б–Њ–∞–≤—В. (2003) —Г—Б—В–∞–љ–Њ–≤–Є–ї–Є —Б–љ–Є–ґ–µ–љ–Є–µ —А–Є—Б–Ї–∞ —А–∞–Ј–≤–Є—В–Є—П –і–µ–Љ–µ–љ—Ж–Є–Є –њ—А–Є –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є–Є –∞–Љ–∞–љ—В–∞–і–Є–љ–∞ –і–ї—П –ї–µ—З–µ–љ–Є—П –С–Я [23]. –Р–љ–∞–ї–Њ–≥–Є—З–љ—Л–µ –і–∞–љ–љ—Л–µ –њ–Њ–ї—Г—З–µ–љ—Л –і–ї—П –Љ–µ–Љ–∞–љ—В–Є–љ–∞, –Є–Љ–µ—О—Й–µ–≥–Њ –њ–Њ—Е–Њ–ґ–Є–є –Љ–µ—Е–∞–љ–Є–Ј–Љ –і–µ–є—Б—В–≤–Є—П –Є¬†–і–µ–Љ–Њ–љ—Б—В—А–Є—А—Г—О—Й–µ–≥–Њ –њ–Њ–Ј–Є—В–Є–≤–љ–Њ–µ –і–µ–є—Б—В–≤–Є–µ –љ–∞¬†–Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л–µ —Д—Г–љ–Ї—Ж–Є–Є —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–і–µ–Љ–µ–љ—Ж–Є–µ–є –љ–∞¬†—Д–Њ–љ–µ –С–Я –Є¬†–±–Њ–ї–µ–Ј–љ–Є –і–Є—Д—Д—Г–Ј–љ—Л—Е —В–µ–ї–µ—Ж –Ы–µ–≤–Є (–і–µ–Љ–µ–љ—Ж–Є–Є —Б¬†—В–µ–ї—М—Ж–∞–Љ–Є –Ы–µ–≤–Є) [10,¬†24].
–Я—А–µ–і–њ–Њ–ї–∞–≥–∞–ї–Њ—Б—М, —З—В–Њ —Б–љ–Є–ґ–µ–љ–Є–µ —Г—А–Њ–≤–љ—П –њ–Њ–ї–Њ–≤—Л—Е¬†–≥–Њ—А–Љ–Њ–љ–Њ–≤ –≤¬†–њ–Њ—Б—В¬≠–Љ–µ–љ–Њ–њ–∞—Г–Ј–∞–ї—М–љ–Њ–Љ –њ–µ—А–Є–Њ–і–µ –Љ–Њ–ґ–µ—В –≤–ї–Є—П—В—М –љ–∞¬†—Б–Ї–Њ—А–Њ—Б—В—М —А–∞–Ј–≤–Є—В–Є—П –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л—Е –љ–∞—А—Г—И–µ–љ–Є–є. –Э–∞–њ—А–Є–Љ–µ—А, –Ј–∞–Љ–µ—Б—В–Є—В–µ–ї—М–љ–∞—П —В–µ—А–∞–њ–Є—П —Н—Б—В—А–Њ–≥–µ–љ–∞–Љ–Є —Б–њ–Њ—Б–Њ–±—Б—В–≤–Њ–≤–∞–ї–∞ —Б–љ–Є–ґ–µ–љ–Є—О —А–Є—Б–Ї–∞ –і–µ–Љ–µ–љ—Ж–Є–Є.
–Ь–љ–Њ–≥–Њ–Ї—А–∞—В–љ–Њ –Њ—Ж–µ–љ–Є–≤–∞–ї–Њ—Б—М –≤–ї–Є—П–љ–Є–µ –њ—А–Є–µ–Љ–∞ —Б—В–∞—В–Є–љ–Њ–≤ –љ–∞¬†—З–∞—Б—В–Њ—В—Г —А–∞–Ј–≤–Є—В–Є—П –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л—Е —А–∞—Б—Б—В—А–Њ–є—Б—В–≤. –С–Њ–ї—М—И–Њ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ, –њ–Њ—Б–≤—П—Й–µ–љ–љ–Њ–µ –Є–Ј—Г—З–µ–љ–Є—О –≤–Ј–∞–Є–Љ–Њ—Б–≤—П–Ј–Є –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л—Е –љ–∞—А—Г—И–µ–љ–Є–є –њ—А–Є –С–Я –Є¬†–њ—А–Є–µ–Љ–∞ —Б—В–∞—В–Є–љ–Њ–≤, –њ–Њ–Ї–∞–Ј–∞–ї–Њ, —З—В–Њ –њ—А–Є–Љ–µ–љ–µ–љ–Є–µ —Б–Є–Љ–≤–∞—Б—В–∞—В–Є–љ–∞ –Љ–Њ–ґ–µ—В —Б–љ–Є–Ј–Є—В—М —А–Є—Б–Ї¬†—А–∞–Ј–≤–Є—В–Є—П –і–µ–Љ–µ–љ—Ж–Є–Є –њ—А–Є –С–Я –±–Њ–ї–µ–µ —З–µ–Љ –љ–∞¬†50% [25]. –С–ї–∞–≥–Њ–њ—А–Є—П—В–љ—Л–є —Н—Д—Д–µ–Ї—В —Б—В–∞—В–Є–љ–Њ–≤ –Љ–Њ–ґ–µ—В –±—Л—В—М –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ –љ–µ¬†—В–Њ–ї—М–Ї–Њ –≤–ї–Є—П–љ–Є–µ–Љ –љ–∞¬†—Б–Њ–њ—Г—В—Б—В–≤—Г—О—Й–Є–µ —Б–Њ—Б—Г–і–Є—Б—В—Л–µ —Д–∞–Ї—В–Њ—А—Л —А–Є—Б–Ї–∞, –љ–Њ¬†–Є –±–ї–Њ–Ї–Є—А–Њ–≤–∞–љ–Є–µ–Љ –≤–Њ–і–Њ—А–Њ–і-–њ–Њ—В—А–µ–±–ї—П—О—Й–µ–є –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –Љ–µ—В–∞–љ–Њ–≥–µ–љ–љ–Њ–є –Љ–Є–Ї—А–Њ—Д–ї–Њ—А—Л –Ї–Є—И–µ—З–љ–Є–Ї–∞ (–±–µ–Ј —Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–Є—П –і–Є—Б–±–Є–Њ–Ј–∞) –Є¬†–Є–љ–≥–Є–±–Є—А—Г—О—Й–Є–Љ –≤–ї–Є—П–љ–Є–µ–Љ –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ–Њ–≥–Њ –≤–Њ–і–Њ—А–Њ–і–∞ –љ–∞¬†—Н–Ї—Б–њ—А–µ—Б—Б–Є—О –Љ–†–Э–Ъ-–Є–љ–і—Г—Ж–Є–±–µ–ї—М–љ–Њ–є NO-—Б–Є–љ—В–∞–Ј—Л –Є¬†–њ—А–Њ–≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л—Е —Ж–Є—В–Њ–Ї–Є–љ–Њ–≤ (–Є–љ—В–µ—А–ї–µ–є–Ї–Є–љ–∞ 1b, 6, —Д–∞–Ї—В–Њ—А–∞ –љ–µ–Ї—А–Њ–Ј–∞ –Њ–њ—Г—Е–Њ–ї–Є –∞–ї—М—Д–∞) [21].
–Ь–Њ—А—Д–Њ–ї–Њ–≥–Є—П
–Т¬†–љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П –±–Њ–ї–µ–Ј–љ—М –Я–∞—А–Ї–Є–љ—Б–Њ–љ–∞ –Њ—В–љ–Њ—Б—П—В –Ї¬†–Ї–ї–∞—Б—Б—Г –Ї–Њ–љ—Д–Њ—А–Љ–∞—Ж–Є–Њ–љ–љ—Л—Е –±–Њ–ї–µ–Ј–љ–µ–є –Љ–Њ–Ј–≥–∞. –≠—В–Є –±–Њ–ї–µ–Ј–љ–Є –≤—Л–Ј–≤–∞–љ—Л –љ–∞—А—Г—И–µ–љ–Є–µ–Љ –Ї–Њ–љ—Д–Њ—А–Љ–∞—Ж–Є–Є –Є¬†–≤–љ—Г—В—А–Є–Ї–ї–µ—В–Њ—З–љ–Њ–≥–Њ –њ—А–Њ—Ж–µ—Б—Б–Є–љ–≥–∞ –Њ–њ—А–µ–і–µ–ї–µ–љ–љ–Њ–≥–Њ –±–µ–ї–Ї–∞, –Ї–Њ—В–Њ—А–Њ–µ –њ—А–Є–≤–Њ–і–Є—В –Ї¬†—Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–Є—О –±–µ–ї–Ї–Њ–≤—Л—Е –∞–≥—А–µ–≥–∞—В–Њ–≤, –Є–љ–Є—Ж–Є–Є—А—Г—О—Й–Є—Е –њ—А–Њ—Ж–µ—Б—Б –љ–µ–є—А–Њ–і–µ–≥–µ–љ–µ—А–∞—Ж–Є–Є. –Т¬†–Њ—Б–љ–Њ–≤–µ –Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ—Л—Е –Љ–µ—Е–∞–љ–Є–Ј–Љ–Њ–≤ –С–Я¬†вАУ –љ–∞—А—Г—И–µ–љ–Є–µ —Д–Њ–ї–і–Є–љ–≥–∞ –±–µ–ї–Ї–∞ –∞–ї—М—Д–∞-—Б–Є–љ—Г–Ї–ї–µ–Є–љ–∞ –≤¬†—Б–Њ—З–µ—В–∞–љ–Є–Є —Б¬†–µ–≥–Њ –њ–Њ–ї–Є–Љ–µ—А–Є–Ј–∞—Ж–Є–µ–є [16].
–Р–ї—М—Д–∞-—Б–Є–љ—Г–Ї–ї–µ–Є–љ¬†вАУ –њ—А–µ—Б–Є–љ–∞–њ—В–Є—З–µ—Б–Ї–Є–є –±–µ–ї–Њ–Ї, —Г—З–∞—Б—В–≤—Г—О—Й–Є–є –≤¬†–≤–µ–Ј–Є–Ї—Г–ї—П—А–љ–Њ–Љ –љ–µ–є—А–Њ–љ–∞–ї—М–љ–Њ–Љ —В—А–∞–љ—Б–њ–Њ—А—В–µ. –Т¬†–Ї–ї–µ—В–Ї–µ –∞–ї—М—Д–∞-—Б–Є–љ—Г–Ї–ї–µ–Є–љ —Б—Г—Й–µ—Б—В–≤—Г–µ—В –≤¬†–Љ–µ–Љ–±—А–∞–љ—Б–≤—П–Ј–∞–љ–љ–Њ–є –Є¬†–љ–∞—В–Є–≤–љ–Њ–є —Д–Њ—А–Љ–∞—Е. –°–≤—П–Ј—Л–≤–∞–љ–Є–µ –∞–ї—М—Д–∞-—Б–Є–љ—Г–Ї–ї–µ–Є–љ–∞ —Б¬†–Љ–µ–Љ–±—А–∞–љ–∞–Љ–Є —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞–µ—В—Б—П –µ–≥–Њ –њ–µ—А–µ—Е–Њ–і–Њ–Љ –≤¬†–∞–ї—М—Д–∞-—Б–њ–Є—А–∞–ї—М. –Т¬†–љ–∞—В–Є–≤–љ–Њ–є —Д–Њ—А–Љ–µ –Њ–љ –њ—А–µ–і—Б—В–∞–≤–ї—П–µ—В —Б–Њ–±–Њ–є —А–∞—Б—В–≤–Њ—А–Є–Љ—Л–є –±–µ–ї–Њ–Ї —Б–Њ¬†—Б–ї–∞–±–Њ —Г–њ–Њ—А—П–і–Њ—З–µ–љ–љ–Њ–є —Б—В—А—Г–Ї—В—Г—А–Њ–є. –Я—А–Є –њ–Њ–≤—Л—И–µ–љ–љ–Њ–є –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є –∞–ї—М—Д–∞-—Б–Є–љ—Г–Ї–ї–µ–Є–љ–∞ –≤¬†—А–∞—Б—В–≤–Њ—А–µ –Њ–±—А–∞–Ј—Г—О—В—Б—П —Д–Є–±—А–Є–ї–ї—Л –Є¬†–∞–≥—А–µ–≥–∞—В—Л, –≤¬†—З–∞—Б—В–љ–Њ—Б—В–Є –њ—А–Њ—Ж–µ—Б—Б –∞–≥—А–µ–≥–∞—Ж–Є–Є —Д–Є–±—А–Є–ї–ї —В–∞—Г-–±–µ–ї–Ї–∞ —В–Є–њ–Є—З–µ–љ –і–ї—П –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є—П —В–µ–ї–µ—Ж –Ы–µ–≤–Є [26]. –Р–≥—А–µ–≥–∞—В—Л –∞–ї—М—Д–∞-—Б–Є–љ—Г–Ї–ї–µ–Є–љ–∞ –љ–∞—А—Г—И–∞—О—В —А–∞–±–Њ—В—Г –њ—А–Њ—В–µ–Њ—Б–Њ–Љ –Є¬†–ї–Є–Ј–Њ—Б–Њ–Љ, —Б—В–Є–Љ—Г–ї–Є—А—Г—П –і–∞–ї—М–љ–µ–є—И–µ–µ –љ–∞–Ї–Њ–њ–ї–µ–љ–Є–µ –Є–Ј–Љ–µ–љ–µ–љ–љ–Њ–≥–Њ –±–µ–ї–Ї–∞ [24].
–Х—Б—В—М –і–∞–љ–љ—Л–µ –Њ¬†–њ—А–Є–Њ–љ–Њ–њ–Њ–і–Њ–±–љ—Л—Е —Б–≤–Њ–є—Б—В–≤–∞—Е –∞–ї—М—Д–∞-—Б–Є–љ—Г–Ї–ї–µ–Є–љ–∞. –Я–Њ–Ї–∞–Ј–∞–љ–∞ —Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В—М –∞–≥—А–µ–≥–∞—В–Њ–≤ –∞–ї—М—Д–∞-—Б–Є–љ—Г–Ї–ї–µ–Є–љ–∞ –Ї¬†—Б–µ–Ї—А–µ—Ж–Є–Є —Б¬†–њ–Њ—Б–ї–µ–і—Г—О—Й–Є–Љ –Ј–∞—Е–≤–∞—В–Њ–Љ —Б–Њ—Б–µ–і–љ–Є–Љ–Є –Ї–ї–µ—В–Ї–∞–Љ–Є –Є¬†–њ–Њ—Б—В–µ–њ–µ–љ–љ—Л–Љ —А–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–Є–µ–Љ –њ–∞—В–Њ–ї–Њ–≥–Є–Є –њ–Њ¬†–љ–µ–є—А–Њ–љ–љ—Л–Љ –њ—Г—В—П–Љ [27].
–Э. Braak (2002) –њ—А–µ–і–њ–Њ–ї–Њ–ґ–Є–ї, —З—В–Њ –њ—А–Є –С–Я –Є–Љ–µ–µ—В –Љ–µ—Б—В–Њ –≤–Њ—Б—Е–Њ–і—П—Й–Є–є —В–Є–њ –њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–≥–Њ –љ–µ–є—А–Њ–і–µ–≥–µ–љ–µ—А–∞—В–Є–≤–љ–Њ–≥–Њ –њ—А–Њ—Ж–µ—Б—Б–∞, –Ї–Њ—В–Њ—А—Л–є –њ—А–Њ—Е–Њ–і–Є—В —И–µ—Б—В—М —Б—В–∞–і–Є–є:
- –Њ—В–ї–Њ–ґ–µ–љ–Є–µ –∞–ї—М—Д–∞-—Б–Є–љ—Г–Ї–ї–µ–Є–љ–∞ –≤¬†–њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Є—Е –≤–µ–≥–µ—В–∞—В–Є–≤–љ—Л—Е¬†–≥–∞–љ–≥–ї–Є—П—Е –Ї–Є—И–µ—З–љ–Є–Ї–∞ –Є¬†–Ї–Њ–ґ–Є;
- –≤–Њ–≤–ї–µ—З–µ–љ–Є–µ —Б—В–≤–Њ–ї–Њ–≤—Л—Е —П–і–µ—А–љ—Л—Е —Б—В—А—Г–Ї—В—Г—А (–Љ–∞–≥–љ–Њ—Ж–µ–ї–ї—О–ї—П—А–љ—Л–µ —З–∞—Б—В–Є —А–µ—В–Є–Ї—Г–ї—П—А–љ–Њ–є —Д–Њ—А–Љ–∞—Ж–Є–Є,¬†–≥–Њ–ї—Г–±–Њ–µ –њ—П—В–љ–Њ);
- –њ–Њ—А–∞–ґ–µ–љ–Є–µ –Ї–Њ–Љ–њ–∞–Ї—В–љ–Њ–є —З–∞—Б—В–Є —З–µ—А–љ–Њ–є —Б—Г–±—Б—В–∞–љ—Ж–Є–Є,¬†–≥–і–µ –њ—А–Њ–Є—Б—Е–Њ–і–Є—В –њ–Њ—В–µ—А—П 80вАУ85% –њ–Є–≥–Љ–µ–љ—В–Є—А–Њ–≤–∞–љ–љ—Л—Е –і–Њ—Д–∞–Љ–Є–љ–µ—А–≥–Є—З–µ—Б–Ї–Є—Е –љ–µ–є—А–Њ–љ–Њ–≤, –∞¬†—В–∞–Ї–ґ–µ –љ–µ–є—А–Њ–і–µ–≥–µ–љ–µ—А–∞—В–Є–≤–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –≤¬†–њ–µ–і—Г–љ–Ї—Г–ї–Њ–њ–Њ–љ—В–Є–љ–љ–Њ–Љ —П–і—А–µ, –Њ—А–∞–ї—М–љ–Њ–Љ —П–і—А–µ —И–≤–∞, —Е–Њ–ї–Є–љ–µ—А–≥–Є—З–µ—Б–Ї–Є—Е –Љ–∞–≥–љ–Њ—Ж–µ–ї–ї—О–ї—П—А–љ—Л—Е —П–і—А–∞—Е –±–∞–Ј–∞–ї—М–љ—Л—Е –Њ—В–і–µ–ї–Њ–≤ –њ–µ—А–µ–і–љ–µ–≥–Њ –Љ–Њ–Ј–≥–∞ (–≤¬†—В–Њ–Љ —З–Є—Б–ї–µ –≤¬†–±–∞–Ј–∞–ї—М–љ–Њ–Љ —П–і—А–µ –Ь–µ–є–љ–µ—А—В–∞), —В—Г–±–µ—А–Њ–Љ–∞–Љ–Љ–Є–ї—П—А–љ–Њ–Љ —П–і—А–µ¬†–≥–Є–њ–Њ—В–∞–ї–∞–Љ—Г—Б–∞;
- –њ–Њ—А–∞–ґ–µ–љ–Є–µ –≤–Є—Б–Њ—З–љ–Њ–≥–Њ –Љ–µ–Ј–Њ¬≠–Ї–Њ—А—В–µ–Ї—Б–∞ (–њ—А–µ–і–њ–Њ–ї–Њ–ґ–Є—В–µ–ї—М–љ–Њ, –≤–Ї–ї—О—З–∞—П –ї–Є–Љ–±–Є—З–µ—Б–Ї—Г—О —Б–Є—Б—В–µ–Љ—Г) –Є¬†–≥–Є–њ–њ–Њ–Ї–∞–Љ–њ–∞;
- –≤–Њ–≤–ї–µ—З–µ–љ–Є–µ –∞—Б—Б–Њ—Ж–Є–∞—В–Є–≤–љ—Л—Е –Ј–Њ–љ –њ—А–µ—Д—А–Њ–љ—В–∞–ї—М–љ–Њ–є, –≤–Є—Б–Њ—З–љ–Њ–є, —В–µ–Љ–µ–љ–љ–Њ–є, –Ј–∞—В—Л–ї–Њ—З–љ–Њ–є –Ї–Њ—А—Л;
- –њ–Њ—А–∞–ґ–µ–љ–Є–µ –њ–µ—А–≤–Є—З–љ—Л—Е –Љ–Њ—В–Њ—А–љ—Л—Е –Є¬†—Б–µ–љ—Б–Њ—А–љ—Л—Е –Ј–Њ–љ –Ї–Њ—А—Л –±–Њ–ї—М—И–Є—Е –њ–Њ–ї—Г—И–∞—А–Є–є¬†–≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ [28].
–°–Њ–≥–ї–∞—Б–љ–Њ H. Hall –Є¬†—Б–Њ–∞–≤—В. (2014), —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–С–Я –Є¬†–і–µ–Љ–µ–љ—Ж–Є–µ–є –і–Њ—Б—В–Њ–≤–µ—А–љ–Њ —З–∞—Й–µ –Њ–±–љ–∞—А—Г–ґ–Є–≤–∞–ї–Є—Б—М –љ–∞–Ї–Њ–њ–ї–µ–љ–Є—П –∞–ї—М—Д–∞-—Б–Є–љ—Г–Ї–ї–µ–Є–љ–∞ –≤¬†–±–∞–Ј–∞–ї—М–љ–Њ–Љ –њ–µ—А–µ–і–љ–µ–Љ –Љ–Њ–Ј–≥–µ –Є¬†–≥–Є–њ–њ–Њ–Ї–∞–Љ–њ–µ –њ–Њ¬†—Б—А–∞–≤–љ–µ–љ–Є—О —Б¬†–њ–∞—Ж–Є–µ–љ—В–∞–Љ–Є —Б¬†–С–Я –±–µ–Ј –і–µ–Љ–µ–љ—Ж–Є–Є. –Р–≤—В–Њ—А—Л –њ—А–µ–і–њ–Њ–ї–∞–≥–∞—О—В, —З—В–Њ –љ–∞–Ї–Њ–њ–ї–µ–љ–Є–µ –∞–ї—М—Д–∞-—Б–Є–љ—Г–Ї–ї–µ–Є–љ–∞ —Б–≤—П–Ј–∞–љ–Њ —Б¬†–љ–µ–є—А–Њ–љ–∞–ї—М–љ–Њ–є –і–Є—Б—Д—Г–љ–Ї—Ж–Є–µ–є –Є¬†—А–∞–Ј–≤–Є—В–Є–µ–Љ –Ј–љ–∞—З–Є—В–µ–ї—М–љ—Л—Е –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л—Е —А–∞—Б—Б—В—А–Њ–є—Б—В–≤ [29]. –Ф—А—Г–≥–Є–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞—В–µ–ї–Є —Б—З–Є—В–∞—О—В, —З—В–Њ –Ї¬†–і–µ–Љ–µ–љ—Ж–Є–Є –њ—А–Є –С–Я –њ—А–Є–≤–Њ–і—П—В –Њ—В–ї–Њ–ґ–µ–љ–Є–µ —В–µ–ї–µ—Ж –Ы–µ–≤–Є –Є¬†–љ–µ–є—А–Њ–і–µ–≥–µ–љ–µ—А–∞—Ж–Є—П –ї–Є–Љ–±–Є—З–µ—Б–Ї–Є—Е –Є¬†–Ї–Њ—А—В–Є–Ї–∞–ї—М–љ—Л—Е –Ј–Њ–љ [30], –≤¬†—З–∞—Б—В–љ–Њ—Б—В–Є –Ї–Њ—А—Л –ї–Њ–±–љ—Л—Е –і–Њ–ї–µ–є, –њ–Њ—П—Б–љ–Њ–є –Є–Ј–≤–Є–ї–Є–љ—Л [31], –∞¬†—В–∞–Ї–ґ–µ –Ї–Њ—А—Л –≤–Є—Б–Њ—З–љ—Л—Е –і–Њ–ї–µ–є¬†–≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ [32].
–Ь–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–є —Б—Г–±—Б—В—А–∞—В –і–µ–Љ–µ–љ—Ж–Є–Є –њ—А–Є –С–Я¬†вАУ –њ—А–µ–і–Љ–µ—В –Њ–ґ–Є–≤–ї–µ–љ–љ—Л—Е –і–Є—Б–Ї—Г—Б—Б–Є–є. –Э–∞–Ї–Њ–њ–ї–µ–љ–Њ –Љ–љ–Њ–≥–Њ –і–∞–љ–љ—Л—Е, —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г—О—Й–Є—Е –Њ–±¬†–Њ–±–љ–∞—А—Г–ґ–µ–љ–Є–Є –њ—А–Є –∞—Г—В–Њ–њ—Б–Є–Є —Г¬†—З–∞—Б—В–Є –њ–∞—Ж–Є–µ–љ—В–Њ–≤ (–Њ—В¬†17,6% [33] –і–Њ 40%) —Б¬†–С–Я –Є¬†–і–µ–Љ–µ–љ—Ж–Є–µ–є –њ–Њ–Љ–Є–Љ–Њ —В–µ–ї–µ—Ж –Ы–µ–≤–Є –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ–≥–Њ –Ї–Њ–ї–Є—З–µ—Б—В–≤–∞ –∞–Љ–Є–ї–Њ–Є–і–љ—Л—Е –±–ї—П—И–µ–Ї –Є¬†–љ–µ–є—А–Њ—Д–Є–±—А–Є–ї–ї—П—А–љ—Л—Е —Б–њ–ї–µ—В–µ–љ–Є–є –≤¬†—Б—В—А–Є–∞—В—Г–Љ–µ, –Ї–Њ—А–µ¬†–≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞, –≤¬†—В–Њ–Љ —З–Є—Б–ї–µ –≤¬†–њ–Њ—П—Б–љ–Њ–є –Є–Ј–≤–Є–ї–Є–љ–µ,¬†вАУ —В–Є–њ–Є—З–љ—Л—Е –і–ї—П –±–Њ–ї–µ–Ј–љ–Є –Р–ї—М—Ж–≥–µ–є–Љ–µ—А–∞ –Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е –Є–Ј–Љ–µ–љ–µ–љ–Є–є [30, 34]. –Э. Hall –Є¬†—Б–Њ–∞–≤—В. (2014), –Ь. Hely –Є¬†—Б–Њ–∞–≤—В. (2008) —Б—З–Є—В–∞—О—В, —З—В–Њ —Б–Њ—З–µ—В–∞–љ–Є–µ –њ–∞—В–Њ–ї–Њ–≥–Є–Є –∞–Љ–Є–ї–Њ–Є–і–љ–Њ–≥–Њ –±–µ–ї–Ї–∞ –Є¬†—Б–Є–љ—Г–Ї–ї–µ–Є–љ–∞ –Љ–Њ–ґ–µ—В –њ—А–Є–≤–Њ–і–Є—В—М –Ї¬†—Г—Б–Ї–Њ—А–µ–љ–Є—О¬†–≥–Є–±–µ–ї–Є –Ї–ї–µ—В–Њ–Ї, –≤—Б–ї–µ–і—Б—В–≤–Є–µ —З–µ–≥–Њ –і–µ–Љ–µ–љ—Ж–Є—П —А–∞–Ј–≤–Є–≤–∞–µ—В—Б—П –љ–∞¬†—А–∞–љ–љ–µ–є —Б—В–∞–і–Є–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П [30, 33, 35].
–Т¬†–ї–Є—В–µ—А–∞—В—Г—А–µ –µ—Б—В—М –µ–і–Є–љ–Є—З–љ—Л–µ —Б–Њ–Њ–±—Й–µ–љ–Є—П –Њ–±¬†–Њ–±–љ–∞—А—Г–ґ–µ–љ–Є–Є –њ—А–Є –С–Я —Б¬†–і–µ–Љ–µ–љ—Ж–Є–µ–є –Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е –Є–Ј–Љ–µ–љ–µ–љ–Є–є, —Е–∞—А–∞–Ї—В–µ—А–љ—Л—Е –і–ї—П –±–Њ–ї–µ–Ј–љ–Є –Я–Є–Ї–∞: –Љ–Є–Ї—А–Њ–≤–∞–Ї—Г–Њ–ї–Є–Ј–∞—Ж–Є—П –љ–µ–є—А–Њ–љ–Њ–≤ –Є¬†–±–∞–ї–ї–Њ–љ–Њ–Њ–±—А–∞–Ј–љ—Л–µ –Ї–ї–µ—В–Ї–Є –≤–Њ —Д—А–Њ–љ—В–Њ–ї–∞—В–µ—А–∞–ї—М–љ—Л—Е –Є¬†–Њ—А–±–Є—В–Њ—Д—А–Њ–љ—В–∞–ї—М–љ—Л—Е –Ї–Њ—А—В–Є–Ї–∞–ї—М–љ—Л—Е –Њ–±–ї–∞—Б—В—П—Е –Є¬†–њ–Њ—П—Б–љ–Њ–є –Є–Ј–≤–Є–ї–Є–љ–µ, —В–∞—Г-–њ–Њ–Ј–Є—В–Є–≤–љ—Л–µ —Ж–Є—В–Њ¬≠–њ–ї–∞–Ј–Љ–∞—В–Є—З–µ—Б–Ї–Є–µ –љ–µ–є—А–Њ–љ–∞–ї—М–љ—Л–µ –≤–Ї–ї—О—З–µ–љ–Є—П. –°–ї–µ–і—Г–µ—В –Њ—В–Љ–µ—В–Є—В—М, —З—В–Њ –≤–Њ–Ј–Љ–Њ–ґ–љ—Л–є –≤–Ї–ї–∞–і –љ–µ–є—А–Њ–і–µ–≥–µ–љ–µ—А–∞—В–Є–≤–љ—Л—Е –Є–Ј–Љ–µ–љ–µ–љ–Є–є –њ–Њ¬†—В–Є–њ—Г –ї–Њ–±–љ–Њ-–≤–Є—Б–Њ—З–љ–Њ–є –і–µ–≥–µ–љ–µ—А–∞—Ж–Є–Є –њ—А–Є –С–Я —Б¬†–і–µ–Љ–µ–љ—Ж–Є–µ–є —П–≤–љ–Њ –Є–Ј—Г—З–µ–љ –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ [36].
–£¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–і–µ–Љ–µ–љ—Ж–Є–µ–є –Є¬†–С–Я –≤¬†—Е–Њ–і–µ –Љ–∞–≥–љ–Є—В–љ–Њ-—А–µ–Ј–Њ–љ–∞–љ—Б–љ–Њ–є —В–Њ–Љ–Њ–≥—А–∞—Д–Є–Є (–Ь–†–Ґ)¬†–≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ –љ–µ—А–µ–і–Ї–Њ –Њ–±–љ–∞—А—Г–ґ–Є–≤–∞—О—В—Б—П –і–Є—Д—Д—Г–Ј–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –±–µ–ї–Њ–≥–Њ –≤–µ—Й–µ—Б—В–≤–∞ –≤¬†–≤–Є–і–µ –њ–µ—А–Є–≤–µ–љ—В—А–Є–Ї—Г–ї—П—А–љ–Њ–≥–Њ –Є¬†—Б—Г–±–Ї–Њ—А—В–Є–Ї–∞–ї—М–љ–Њ–≥–Њ –ї–µ–є–Ї–Њ–∞—А–µ–Њ–Ј–∞. –Ъ–∞–Ї –њ—А–∞–≤–Є–ї–Њ, –Њ–±—И–Є—А–љ–Њ—Б—В—М –Є¬†–ї–Њ–Ї–∞–ї–Є–Ј–∞—Ж–Є—П –і–∞–љ–љ—Л—Е –Є–Ј–Љ–µ–љ–µ–љ–Є–є –љ–µ–≤–µ–ї–Є–Ї–Є –Є¬†–љ–µ –Љ–Њ–≥—Г—В –±—Л—В—М —Б—Г–±—Б—В—А–∞—В–Њ–Љ –і–µ–Љ–µ–љ—Ж–Є–Є, –љ–Њ, –њ—А–µ–і–њ–Њ–ї–Њ–ґ–Є—В–µ–ї—М–љ–Њ, –≤–ї–Є—П—О—В –љ–∞¬†—Б–Ї–Њ—А–Њ—Б—В—М —А–∞–Ј–≤–Є—В–Є—П –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л—Е –љ–∞—А—Г—И–µ–љ–Є–є [1].
–Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, –Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –њ—А–Є –С–Я —Б¬†–і–µ–Љ–µ–љ—Ж–Є–µ–є –љ–µ–Њ–і–љ–Њ—А–Њ–і–љ—Л. –°–∞–Љ—Л–є —З–∞—Б—В—Л–є –Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–є –≤–∞—А–Є–∞–љ—В¬†вАУ —А–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–љ–∞—П –љ–µ–є—А–Њ–і–µ–≥–µ–љ–µ—А–∞—Ж–Є—П —Ж–µ—А–µ–±—А–∞–ї—М–љ—Л—Е —Б—В—А—Г–Ї—В—Г—А —Б¬†–і–Є—Д—Д—Г–Ј–љ—Л–Љ –Њ—В–ї–Њ–ґ–µ–љ–Є–µ–Љ —В–µ–ї–µ—Ж –Ы–µ–≤–Є. –Ф–ї—П —А–∞–Ј–≤–Є—В–Є—П –і–µ–Љ–µ–љ—Ж–Є–Є –≤–∞–ґ–љ–Њ –њ–Њ—А–∞–ґ–µ–љ–Є–µ¬†–≥–Є–њ–њ–Њ–Ї–∞–Љ–њ–∞, –ї–Є–Љ–±–Є—З–µ—Б–Ї–Њ–є —Б–Є—Б—В–µ–Љ—Л –Є¬†–Ї–Њ—А—Л –±–Њ–ї—М—И–Є—Е –њ–Њ–ї—Г—И–∞—А–Є–є¬†–≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞. –£¬†40% –њ–∞—Ж–Є–µ–љ—В–Њ–≤, —Б—В—А–∞–і–∞—О—Й–Є—Е –С–Я –Є¬†–і–µ–Љ–µ–љ—Ж–Є–µ–є, —В–Є–њ–Є—З–љ–∞—П –і–ї—П –С–Я –Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–∞—П –Ї–∞—А—В–Є–љ–∞ —Б–Њ—З–µ—В–∞–µ—В—Б—П —Б¬†–Є–Ј–Љ–µ–љ–µ–љ–Є—П–Љ–Є, —Е–∞—А–∞–Ї—В–µ—А–љ—Л–Љ–Є –і–ї—П –±–Њ–ї–µ–Ј–љ–Є –Р–ї—М—Ж–≥–µ–є–Љ–µ—А–∞. –†–µ–ґ–µ –њ—А–Є –С–Я –Є¬†–і–µ–Љ–µ–љ—Ж–Є–Є –Њ—В–Љ–µ—З–∞–µ—В—Б—П –Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–∞—П –Ї–∞—А—В–Є–љ–∞ –ї–Њ–±–љ–Њ-–≤–Є—Б–Њ—З–љ–Њ–є –і–µ–≥–µ–љ–µ—А–∞—Ж–Є–Є. –Т–Њ–Ј–Љ–Њ–ґ–љ–Њ —В–∞–Ї–ґ–µ —Б–Њ—З–µ—В–∞–љ–Є–µ –Є—И–µ–Љ–Є—З–µ—Б–Ї–Њ–≥–Њ –Є¬†–љ–µ–є—А–Њ–і–µ–≥–µ–љ–µ—А–∞—В–Є–≤–љ–Њ–≥–Њ –њ—А–Њ—Ж–µ—Б—Б–∞ —Г¬†–Њ–і–љ–Њ–≥–Њ –њ–∞—Ж–Є–µ–љ—В–∞. –Т¬†—Н—В–Њ–Љ —Б–ї—Г—З–∞–µ —Ж–µ—А–µ–±—А–∞–ї—М–љ–∞—П –Є—И–µ–Љ–Є—П, –≤–µ—А–Њ—П—В–љ–Њ, –±—Г–і–µ—В —Г—Б–Ї–Њ—А—П—В—М —А–∞–Ј–≤–Є—В–Є–µ –љ–µ–є—А–Њ–і–µ–≥–µ–љ–µ—А–∞—Ж–Є–Є.
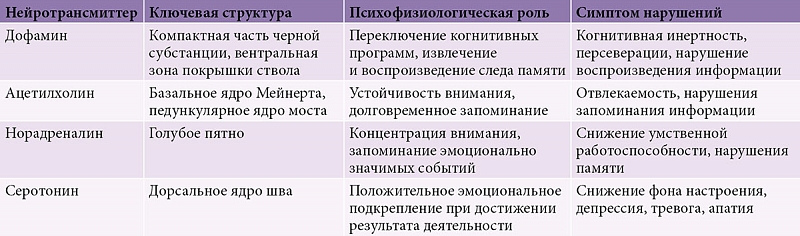
–Э–µ–є—А–Њ–Љ–µ–і–Є–∞—В–Њ—А–љ—Л–µ –љ–∞—А—Г—И–µ–љ–Є—П
–Т¬†–љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П –С–Я —Б—З–Є—В–∞–µ—В—Б—П –Љ—Г–ї—М—В–Є–Љ–µ–і–Є–∞—В–Њ—А–љ—Л–Љ —А–∞—Б—Б—В—А–Њ–є—Б—В–≤–Њ–Љ, —Б–≤—П–Ј–∞–љ–љ—Л–Љ —Б¬†–і–Є—Б—Д—Г–љ–Ї—Ж–Є–µ–є —А–∞–Ј–ї–Є—З–љ—Л—Е –Њ—В–і–µ–ї–Њ–≤ —Ж–µ–љ—В—А–∞–ї—М–љ–Њ–є –Є¬†–њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Њ–є –љ–µ—А–≤–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л (—В–∞–±–ї.¬†1) [37].
–°–љ–Є–ґ–µ–љ–Є–µ —З–Є—Б–ї–µ–љ–љ–Њ—Б—В–Є –і–Њ—Д–∞–Љ–Є–љ–µ—А–≥–Є—З–µ—Б–Ї–Є—Е –љ–µ–є—А–Њ–љ–Њ–≤ –≤¬†–Ї–Њ–Љ–њ–∞–Ї—В¬≠–љ–Њ–є —З–∞—Б—В–Є —З–µ—А–љ–Њ–є —Б—Г–±—Б—В–∞–љ—Ж–Є–Є –њ—А–Є–≤–Њ–і–Є—В –Ї¬†—Г–Љ–µ–љ—М—И–µ–љ–Є—О —Б–Њ–і–µ—А–ґ–∞–љ–Є—П –і–Њ—Д–∞–Љ–Є–љ–∞ –≤¬†–њ–Њ–ї–Њ—Б–∞—В–Њ–Љ —В–µ–ї–µ, —З—В–Њ –≤—Л–Ј—Л–≤–∞–µ—В –і–Є—Б—Д—Г–љ–Ї—Ж–Є—О –љ–µ–є—А–Њ–љ–Њ–≤ –і—А—Г–≥–Є—Е –±–∞–Ј–∞–ї—М–љ—Л—Е¬†–≥–∞–љ–≥–ї–Є–µ–≤, –њ—А–µ–ґ–і–µ –≤—Б–µ–≥–Њ —А–∞—Б—В–Њ—А–Љ–∞–ґ–Є–≤–∞–љ–Є–µ –љ–µ–є—А–Њ–љ–Њ–≤ –≤–љ—Г—В—А–µ–љ–љ–µ–≥–Њ —Б–µ–≥–Љ–µ–љ—В–∞ –±–ї–µ–і–љ–Њ–≥–Њ —И–∞—А–∞ –Є¬†—А–µ—В–Є–Ї—Г–ї—П—А–љ–Њ–є —З–∞—Б—В–Є —З–µ—А–љ–Њ–є —Б—Г–±—Б—В–∞–љ—Ж–Є–Є [38], —В–Њ—А–Љ–Њ–ґ–µ–љ–Є–µ —В–∞–ї–∞–Љ–Њ–Ї–Њ—А—В–Є–Ї–∞–ї—М–љ—Л—Е –љ–µ–є—А–Њ–љ–Њ–≤ –Є¬†–і–µ—Д–Є—Ж–Є—В –∞–Ї—В–Є–≤–∞—Ж–Є–Є –љ–µ–є—А–Њ–љ–Њ–≤ –і–Њ–њ–Њ–ї–љ–Є—В–µ–ї—М–љ–Њ–є –Љ–Њ—В–Њ—А–љ–Њ–є –Ї–Њ—А—Л. –°¬†—Г–Ї–∞–Ј–∞–љ–љ—Л–Љ–Є –Є–Ј–Љ–µ–љ–µ–љ–Є—П–Љ–Є —Б–≤—П–Ј—Л–≤–∞—О—В —А–∞–Ј–≤–Є—В–Є–µ –Њ—Б–љ–Њ–≤–љ—Л—Е –і–≤–Є–≥–∞—В–µ–ї—М–љ—Л—Е —Б–Є–Љ–њ—В–Њ–Љ–Њ–≤ –С–Я –Є¬†–Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л—Е –і–Є–Ј—А–µ–≥—Г–ї—П—В–Њ—А–љ—Л—Е —А–∞—Б—Б—В—А–Њ–є—Б—В–≤ [23, 39]. –Э–µ–є—А–Њ–і–µ–≥–µ–љ–µ—А–∞—В–Є–≤–љ—Л–є –њ—А–Њ—Ж–µ—Б—Б –њ—А–Є –С–Я –њ–Њ—А–∞–ґ–∞–µ—В —В–∞–Ї–ґ–µ –≤–µ–љ—В—А–∞–ї—М–љ—Л–µ –Њ—В–і–µ–ї—Л —Б—А–µ–і–љ–µ–≥–Њ –Љ–Њ–Ј–≥–∞ –Є¬†–≥–Њ–ї—Г–±–Њ–µ –њ—П—В–љ–Њ [40], —З—В–Њ –њ—А–Њ—П–≤–ї—П–µ—В—Б—П –љ–Њ—А–∞–і—А–µ–љ–µ—А–≥–Є—З–µ—Б–Ї–Њ–є –Є¬†–і–Њ—Д–∞–Љ–Є–љ–µ—А–≥–Є—З–µ—Б–Ї–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М—О [29, 40].
–†–µ–Ј—Г–ї—М—В–∞—В –њ–Њ—А–∞–ґ–µ–љ–Є—П –∞—Ж–µ—В–Є–ї—Е–Њ–ї–Є–љ–µ—А–≥–Є—З–µ—Б–Ї–Њ–≥–Њ —П–і—А–∞ –Ь–µ–є–љ–µ—А—В–∞¬†вАУ –∞—Ж–µ—В–Є–ї—Е–Њ–ї–Є–љ–µ—А–≥–Є—З–µ—Б–Ї–Є–є –і–µ—Д–Є—Ж–Є—В –љ–µ–є—А–Њ–љ–Њ–≤ –Ї–Њ—А—Л —В–µ–Љ–µ–љ–љ—Л—Е, –≤–Є—Б–Њ—З–љ—Л—Е –Є¬†–Ј–∞—В—Л–ї–Њ—З–љ—Л—Е –Њ—В–і–µ–ї–Њ–≤ –±–Њ–ї—М—И–Є—Е –њ–Њ–ї—Г—И–∞—А–Є–є¬†–≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞. –°–Њ–≥–ї–∞—Б–љ–Њ –њ—А–Њ–≤–µ–і–µ–љ–љ—Л–Љ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П–Љ, –∞—В—А–Њ—Д–Є—П –Ї–Њ—А—Л –Љ–µ–і–Є–∞–ї—М–љ—Л—Е –Њ—В–і–µ–ї–Њ–≤ –≤–Є—Б–Њ—З–љ—Л—Е –і–Њ–ї–µ–є,¬†–≥–Є–њ–њ–Њ–Ї–∞–Љ–њ–∞ –Є¬†—Б–Њ–њ—Г—В—Б—В–≤—Г—О—Й–Є–є —Н—В–Є–Љ –Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–Љ –Є–Ј–Љ–µ–љ–µ–љ–Є—П–Љ –∞—Ж–µ—В–Є–ї—Е–Њ–ї–Є–љ–µ—А–≥–Є—З–µ—Б–Ї–Є–є –і–µ—Д–Є—Ж–Є—В –Љ–Њ–≥—Г—В –±—Л—В—М –Њ—Б–љ–Њ–≤–∞–љ–Є–µ–Љ –і–ї—П —А–∞–Ј–≤–Є—В–Є—П¬†–≥–Є–њ–њ–Њ–Ї–∞–Љ–њ–∞–ї—М–љ—Л—Е –љ–∞—А—Г—И–µ–љ–Є–є –њ–∞–Љ—П—В–Є, –Њ–±—Л—З–љ–Њ –љ–µ¬†—Е–∞—А–∞–Ї—В–µ—А–љ—Л—Е –і–ї—П –С–Я [29]. –Т¬†—В–Њ –ґ–µ –≤—А–µ–Љ—П –∞—Ж–µ—В–Є–ї—Е–Њ–ї–Є–љ–µ—А–≥–Є—З–µ—Б–Ї–∞—П –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М –њ—А–Є –С–Я –≤—Б–µ –ґ–µ –≤—Л—А–∞–ґ–µ–љ–∞ –Љ–µ–љ—М—И–µ, —З–µ–Љ –њ—А–Є –±–Њ–ї–µ–Ј–љ–Є –Р–ї—М—Ж–≥–µ–є–Љ–µ—А–∞, —З—В–Њ, –њ—А–µ–і–њ–Њ–ї–Њ–ґ–Є—В–µ–ї—М–љ–Њ, –Њ–±—К—П—Б–љ—П–µ—В –ї—Г—З—И–Є–є –Њ—В–≤–µ—В –љ–∞¬†–ї–µ—З–µ–љ–Є–µ –∞—Ж–µ—В–Є–ї—Е–Њ–ї–Є–љ–µ—А–≥–Є—З–µ—Б–Ї–Є–Љ–Є –њ—А–µ–њ–∞—А–∞—В–∞–Љ–Є —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–С–Я –Є¬†–і–µ–Љ–µ–љ—Ж–Є–µ–є, –∞¬†—В–∞–Ї–ґ–µ —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–і–µ–Љ–µ–љ—Ж–Є–µ–є —Б¬†—В–µ–ї—М—Ж–∞–Љ–Є –Ы–µ–≤–Є.
–°–µ—А–Њ—В–Њ–љ–Є–љ–µ—А–≥–Є—З–µ—Б–Ї–Є–µ –і–Њ—А—Б–∞–ї—М–љ—Л–µ —П–і—А–∞ —И–≤–∞ —Б—В–≤–Њ–ї–∞¬†–≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ —В–Њ–ґ–µ –њ–Њ–і–≤–µ—А–ґ–µ–љ—Л –љ–µ–є—А–Њ–і–µ–≥–µ–љ–µ—А–∞—В–Є–≤–љ–Њ–Љ—Г –њ—А–Њ—Ж–µ—Б—Б—Г –њ—А–Є –С–Я —Б¬†–і–µ–Љ–µ–љ—Ж–Є–µ–є, –≤¬†—А–µ–Ј—Г–ї—М—В–∞—В–µ —З–µ–≥–Њ —Б–љ–Є–ґ–∞–µ—В—Б—П —Г—А–Њ–≤–µ–љ—М —Б–µ—А–Њ—В–Њ–љ–Є–љ–∞ –≤¬†—Б—В—А–Є–∞—В—Г–Љ–µ –Є¬†–Ї–Њ—А–µ. –°–µ—А–Њ—В–Њ–љ–Є–љ —Г—З–∞—Б—В–≤—Г–µ—В –≤¬†—Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–Є–Є –њ–Њ–ї–Њ–ґ–Є—В–µ–ї—М–љ–Њ–≥–Њ —Н–Љ–Њ—Ж–Є–Њ–љ–∞–ї—М–љ–Њ–≥–Њ –њ–Њ–і–Ї—А–µ–њ–ї–µ–љ–Є—П –њ—А–Є –і–Њ—Б—В–Є–ґ–µ–љ–Є–Є —А–µ–Ј—Г–ї—М—В–∞—В–∞ –і–µ—П—В–µ–ї—М–љ–Њ—Б—В–Є. –°–љ–Є–ґ–µ–љ–Є–µ —Г—А–Њ–≤–љ—П —Б–µ—А–Њ—В–Њ–љ–Є–љ–∞ –≤¬†—Б–Њ–Њ—В–≤–µ—В—Б—В–≤—Г—О—Й–Є—Е –Њ—В–і–µ–ї–∞—Е¬†–≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ –Љ–Њ–ґ–µ—В —Б—В–∞—В—М –њ—А–Є—З–Є–љ–Њ–є —А–∞–Ј–≤–Є—В–Є—П —Н–Љ–Њ—Ж–Є–Њ–љ–∞–ї—М–љ–Њ-–њ–Њ–≤–µ–і–µ–љ—З–µ—Б–Ї–Є—Е –љ–∞—А—Г—И–µ–љ–Є–є (–і–µ–њ—А–µ—Б—Б–Є–Є –Є¬†–∞–њ–∞—В–Є–Є) [39].
–Я–Њ—А–∞–ґ–µ–љ–Є–µ –љ–µ–є—А–Њ–љ–Њ–≤ –≤¬†–≥–Њ–ї—Г–±–Њ–Љ –њ—П—В–љ–µ –≤—Л–Ј—Л–≤–∞–µ—В –љ–µ–і–Њ—Б—В–∞—В–Њ–Ї –љ–Њ—А¬≠–∞–і—А–µ–љ–∞–ї–Є–љ–∞. –Т¬†—Б–≤–Њ—О –Њ—З–µ—А–µ–і—М —Б–љ–Є–ґ–µ–љ–Є–µ —Г—А–Њ–≤–љ—П —Ж–µ—А–µ–±—А–∞–ї—М–љ–Њ–≥–Њ –љ–Њ—А–∞–і—А–µ–љ–∞–ї–Є–љ–∞ –Љ–Њ–ґ–µ—В –њ—А–Є–≤–Њ–і–Є—В—М –Ї¬†–љ–∞—А—Г—И–µ–љ–Є—О –њ—А–Њ—Ж–µ—Б—Б–∞ –Ј–∞–њ–Њ–Љ–Є–љ–∞–љ–Є—П —Н–Љ–Њ—Ж–Є–Њ–љ–∞–ї—М–љ–Њ –Ј–љ–∞—З–Є–Љ—Л—Е —Б–Њ–±—Л—В–Є–є –Є¬†–Ї–Њ–ї–µ–±–∞–љ–Є—О –ї–Є–±–Њ –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є —Г—А–Њ–≤–љ—П –≤–љ–Є–Љ–∞–љ–Є—П [41, 42], —Г–Љ—Б—В–≤–µ–љ–љ–Њ–є —А–∞–±–Њ—В–Њ—Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В–Є, –њ–∞–Љ—П—В–Є [39].
–Ь. Ray –Є¬†I. Bohr (2004) —Г—Б—В–∞–љ–Њ–≤–Є–ї–Є, —З—В–Њ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –≤¬†–љ–Є–Ї–Њ—В–Є–љ–Њ–≤—Л—Е —А–µ—Ж–µ–њ—В–Њ—А–∞—Е –Љ–Њ–≥—Г—В –±—Л—В—М —Б–≤—П–Ј–∞–љ—Л —Б¬†–љ–∞—А—Г—И–µ–љ–Є—П–Љ–Є –≤–љ–Є–Љ–∞–љ–Є—П, –њ–∞–Љ—П—В–Є –Є¬†–Њ–±—Г—Б–ї–Њ–≤–ї–Є–≤–∞—В—М —А–∞–Ј–≤–Є—В–Є–µ –Ј—А–Є—В–µ–ї—М–љ—Л—Е¬†–≥–∞–ї–ї—О—Ж–Є–љ–∞—Ж–Є–є [42]. –Т¬†–Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П—Е, –њ–Њ—Б–≤—П—Й–µ–љ–љ—Л—Е –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є¬†–≥–ї—Г—В–∞–Љ–∞—В–µ—А–≥–Є—З–µ—Б–Ї–Њ–є —Б–Є—Б—В–µ–Љ—Л –њ—А–Є –С–Я —Б¬†–і–µ–Љ–µ–љ—Ж–Є–µ–є, –≤—Л—П–≤–ї–µ–љ–Њ —Б–љ–Є–ґ–µ–љ–Є–µ —Д—Г–љ–Ї—Ж–Є–Є –љ–µ—Б–Ї–Њ–ї—М–Ї–Є—Е¬†–≥–ї—Г—В–∞–Љ–∞—В–љ—Л—Е —А–µ—Ж–µ–њ—В–Њ—А–Њ–≤ [43].
–Ф–Є–∞–≥–љ–Њ—Б—В–Є–Ї–∞
–Ф–Є–∞–≥–љ–Њ—Б—В–Є—З–µ—Б–Ї–Є–µ –Ї—А–Є—В–µ—А–Є–Є
–Я–Њ–і—Е–Њ–і –Ї¬†–і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–µ –і–µ–Љ–µ–љ—Ж–Є–Є –њ—А–Є –С–Я –Љ–µ–љ—П–ї—Б—П, –Њ—Б–Њ–±–µ–љ–љ–Њ –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ –Ј–∞ –њ–Њ—Б–ї–µ–і–љ–Є–µ 20 –ї–µ—В. –Ґ–∞–Ї, –і–Њ 2007¬†–≥.¬†–і–ї—П —Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–Є—П —Н—В–Њ–≥–Њ –і–Є–∞–≥–љ–Њ–Ј–∞ –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–ї–Є—Б—М –Ї—А–Є—В–µ—А–Є–Є –Ь–µ–ґ–і—Г–љ–∞—А–Њ–і–љ–Њ–є –Ї–ї–∞—Б—Б–Є—Д–Є–Ї–∞—Ж–Є–Є –±–Њ–ї–µ–Ј–љ–µ–є 10-–≥–Њ¬†–њ–µ—А–µ—Б–Љ–Њ—В—А–∞ –Є/–Є–ї–Є DSM-IV –Є¬†–і–∞–ґ–µ –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–Є –љ–µ–є—А–Њ–њ—Б–Є—Е–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е —И–Ї–∞–ї (—З–∞—Й–µ –≤—Б–µ–≥–Њ –Ъ—А–∞—В–Ї–Њ–є —И–Ї–∞–ї—Л –Њ—Ж–µ–љ–Ї–Є –њ—Б–Є—Е–Є—З–µ—Б–Ї–Њ–≥–Њ —Б—В–∞—В—Г—Б–∞, –і–µ–Љ–µ–љ—Ж–Є—П —Г—Б—В–∞–љ–∞–≤–ї–Є–≤–∞–ї–∞—Б—М –њ—А–Є —Б—Г–Љ–Љ–µ –±–∞–ї–ї–Њ–≤ 26 –Є–ї–Є –Љ–µ–љ–µ–µ). –Ф–µ–Љ–µ–љ—Ж–Є—П –і–Њ–ї–ґ–љ–∞ –±—Л–ї–∞ —А–∞–Ј–≤–Є—В—М—Б—П –љ–µ¬†—А–∞–љ–µ–µ —З–µ–Љ —З–µ—А–µ–Ј¬†–≥–Њ–і –Њ—В¬†–і–µ–±—О—В–∞ –і–≤–Є–≥–∞—В–µ–ї—М–љ—Л—Е —А–∞—Б—Б—В—А–Њ–є—Б—В–≤. –Т¬†2007¬†–≥.¬†–Ъ–Њ–Љ–Є—В–µ—В —Н–Ї—Б–њ–µ—А—В–Њ–≤ –Ь–µ–ґ–і—Г–љ–∞—А–Њ–і–љ–Њ–≥–Њ –Њ–±—Й–µ—Б—В–≤–∞ —А–∞—Б—Б—В—А–Њ–є—Б—В–≤ –і–≤–Є–ґ–µ–љ–Є—П (Movement Disorder Society Task Force) –Њ–њ—Г–±–ї–Є–Ї–Њ–≤–∞–ї –Ъ–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ –і–Є–∞–≥–љ–Њ—Б—В–Є—З–µ—Б–Ї–Є–µ –Ї—А–Є—В–µ—А–Є–Є –і–µ–Љ–µ–љ—Ж–Є–Є, –∞—Б—Б–Њ—Ж–Є–Є—А–Њ–≤–∞–љ–љ–Њ–є —Б¬†–±–Њ–ї–µ–Ј–љ—М—О –Я–∞—А–Ї–Є–љ—Б–Њ–љ–∞ (—В–∞–±–ї.¬†2).
–Ф–Њ–њ–Њ–ї–љ–Є—В–µ–ї—М–љ—Л–µ –Љ–µ—В–Њ–і—Л –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П
–С–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –Љ–∞—А–Ї–µ—А—Л. –Ъ–Њ–ї–Є¬≠—З–µ—Б—В–≤–µ–љ–љ–Њ –Њ–њ—А–µ–і–µ–ї—П–µ–Љ—Л–µ –±–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –њ–∞—А–∞–Љ–µ—В—А—Л, –Є–љ–і–Є–Ї–∞—В–Њ—А—Л —Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е –Є¬†–њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е –±–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е –њ—А–Њ—Ж–µ—Б—Б–Њ–≤.
–Э–∞–є–і–µ–љ–Њ –љ–µ—Б–Ї–Њ–ї—М–Ї–Њ –њ–Њ—В–µ–љ—Ж–Є–∞–ї—М–љ—Л—Е –±–Є–Њ–Љ–∞—А–Ї–µ—А–Њ–≤, –њ–Њ–Ј–≤–Њ–ї—П—О—Й–Є—Е –≤—Л—П–≤–Є—В—М –љ–µ–є—А–Њ–і–µ–≥–µ–љ–µ—А–∞—В–Є–≤–љ—Л–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П —Б¬†–Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є–µ–Љ —В–µ–ї–µ—Ж –Ы–µ–≤–Є, –≤¬†—В–Њ–Љ —З–Є—Б–ї–µ –С–Я —Б¬†–і–µ–Љ–µ–љ—Ж–Є–µ–є: –∞–ї—М—Д–∞-—Б–Є–љ—Г–Ї–ї–µ–Є–љ, –±–µ—В–∞-–∞–Љ–Є–ї–Њ–Є–і¬†42, —Д–Њ—Б—Д–Њ—А–Є–ї–Є—А–Њ–≤–∞–љ–љ—Л–є —В–∞—Г-–њ—А–Њ—В–µ–Є–љ.
–Р–ї—М—Д–∞-—Б–Є–љ—Г–Ї–ї–µ–Є–љ¬†вАУ –Њ—Б–љ–Њ–≤–љ–Њ–є –±–Є–Њ–Љ–∞—А–Ї–µ—А —Б–Є–љ—Г–Ї–ї–µ–Є–љ–Њ–њ–∞—В–Є–є¬†вАУ –Љ–Њ–ґ–µ—В –±—Л—В—М –≤—Л—П–≤–ї–µ–љ –Є¬†–≤ –Ї—А–Њ–≤–Є, –Є¬†–≤ —Б–њ–Є–љ–љ–Њ–Љ–Њ–Ј–≥–Њ–≤–Њ–є –ґ–Є–і–Ї–Њ—Б—В–Є. –Т¬†–Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П—Е –њ–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ —Г–≤–µ–ї–Є—З–µ–љ–Є–µ —Г—А–Њ–≤–љ—П –Њ–ї–Є–≥–Њ–Љ–µ—А–љ–Њ–≥–Њ –∞–ї—М—Д–∞-—Б–Є–љ—Г–Ї–ї–µ–Є–љ–∞ –≤¬†–ї–Є–Ї–≤–Њ—А–µ –Є¬†–њ–ї–∞–Ј–Љ–µ –Ї—А–Њ–≤–Є –≤—Л—Б–Њ–Ї–Њ —Б–њ–µ—Ж–Є—Д–Є—З–љ–Њ –і–ї—П –С–Я (85% —Б–ї—Г—З–∞–µ–≤). –Ф–∞–ї—М–љ–µ–є—И–Є–є —А–Њ—Б—В –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є –∞–ї—М—Д–∞-—Б–Є–љ—Г–Ї–ї–µ–Є–љ–∞ –≤¬†–њ–ї–∞–Ј–Љ–µ –Ї—А–Њ–≤–Є –њ—А—П–Љ–Њ —Б–≤—П–Ј–∞–љ —Б¬†—А–∞–Ј–≤–Є—В–Є–µ–Љ –і–µ–Љ–µ–љ—Ж–Є–Є. –£¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–С–Я –Є¬†–і–µ–Љ–µ–љ—Ж–Є–µ–є —Г—А–Њ–≤–µ–љ—М –∞–ї—М—Д–∞-—Б–Є–љ—Г–Ї–ї–µ–Є–љ–∞ –≤—Л—И–µ, —З–µ–Љ —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–С–Я –±–µ–Ј –і–µ–Љ–µ–љ—Ж–Є–Є [44]. –Х—Й–µ —Б–њ–µ—Ж–Є—Д–Є—З–љ–µ–µ –њ–Њ–≤—Л—И–µ–љ–Є–µ –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є —Д–Њ—Б—Д–Њ—А–Є–ї–Є—А–Њ–≤–∞–љ–љ–Њ–≥–Њ –∞–ї—М—Д–∞-—Б–Є–љ—Г–Ї–ї–µ–Є–љ–∞. –£¬†–Ј–і–Њ—А–Њ–≤—Л—Е –ї—О–і–µ–є —Д–Њ—Б—Д–Њ—А–Є–ї–Є—А–Њ–≤–∞–љ–Њ –љ–µ¬†–±–Њ–ї–µ–µ 4% –≤—Б–µ–≥–Њ –∞–ї—М—Д–∞-—Б–Є–љ—Г–Ї–ї–µ–Є–љ–∞, —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–С–Я —Н—В–Њ—В –њ–Њ–Ї–∞–Ј–∞—В–µ–ї—М —Б—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ –≤–Њ–Ј—А–∞—Б—В–∞–µ—В [45].
–Я—А–Є —А–∞–Ј–≤–Є—В–Є–Є –љ–µ–є—А–Њ–і–µ–≥–µ–љ–µ—А–∞—Ж–Є–Є —Б¬†–љ–∞–Ї–Њ–њ–ї–µ–љ–Є–µ–Љ –∞–Љ–Є–ї–Њ–Є–і–љ–Њ–≥–Њ –±–µ–ї–Ї–∞ –≤¬†–≤–µ—Й–µ—Б—В–≤–µ¬†–≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ –µ–≥–Њ –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—П –≤¬†—Б–њ–Є–љ–љ–Њ–Љ–Њ–Ј–≥–Њ–≤–Њ–є –ґ–Є–і–Ї–Њ—Б—В–Є —Б–љ–Є–ґ–∞–µ—В—Б—П. –Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–С–Я –Є¬†–±–Њ–ї–µ–Ј–љ—М—О –Р–ї—М—Ж–≥–µ–є–Љ–µ—А–∞ –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є—П –∞–Љ–Є–ї–Њ–Є–і–љ–Њ–≥–Њ –±–µ–ї–Ї–∞ –≤¬†–ї–Є–Ї–≤–Њ—А–µ –±—Г–і–µ—В –љ–Є–ґ–µ —В–∞–Ї–Њ–≤–Њ–є —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–С–Я –±–µ–Ј –і–µ–Љ–µ–љ—Ж–Є–Є –Є¬†–Ј–і–Њ—А–Њ–≤—Л—Е –њ–Њ–ґ–Є–ї—Л—Е –ї—О–і–µ–є [46]. –І–∞—Б—В–Њ —Б–љ–Є–ґ–µ–љ–Є–µ —Г—А–Њ–≤–љ—П –∞–Љ–Є–ї–Њ–Є–і–љ–Њ–≥–Њ –±–µ–ї–Ї–∞ —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞–µ—В—Б—П –і–≤—Г–Љ—П –њ—А–Њ—Ж–µ—Б—Б–∞–Љ–Є. –Я–µ—А–≤—Л–є¬†вАУ —А–µ—Ж–Є–њ—А–Њ–Ї–љ–Њ–µ –њ–Њ–≤—Л—И–µ–љ–Є–µ —Г—А–Њ–≤–љ—П —В–∞—Г-–њ—А–Њ—В–µ–Є–љ–∞, —З—В–Њ —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г–µ—В –Њ¬†–≥–Є–±–µ–ї–Є –љ–µ–є—А–Њ–љ–Њ–≤¬†–≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ –Є¬†–Љ–Њ–ґ–µ—В –Њ—В–Љ–µ—З–∞—В—М—Б—П –њ—А–Є —Ж–µ–ї–Њ–Љ —А—П–і–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є¬†вАУ –Є–љ—Б—Г–ї—М—В–µ, –њ–Њ—Б–ї–µ¬†–≥–µ–љ–µ—А–∞–ї–Є–Ј–Њ–≤–∞–љ–љ–Њ–≥–Њ —Н–њ–Є–ї–µ–њ—В–Є—З–µ—Б–Ї–Њ–≥–Њ –њ—А–Є–њ–∞–і–Ї–∞, –њ—А–Є —Н–љ—Ж–µ—Д–∞–ї–Є—В–µ, –љ–µ–є—А–Њ–і–µ–≥–µ–љ–µ—А–∞—Ж–Є–Є –Є¬†—В.–і. –Т—В–Њ—А–Њ–є¬†вАУ –њ–Њ—П–≤–ї–µ–љ–Є–µ –≤¬†—Б–њ–Є–љ–љ–Њ–Љ–Њ–Ј–≥–Њ–≤–Њ–є –ґ–Є–і–Ї–Њ—Б—В–Є¬†–≥–Є–њ–µ—А—Д–Њ—Б—Д–Њ—А–Є–ї–Є—А–Њ–≤–∞–љ–љ–Њ–≥–Њ —В–∞—Г-–±–µ–ї–Ї–∞, —З—В–Њ –≤¬†—Б–Њ—З–µ—В–∞–љ–Є–Є —Б–Њ¬†—Б–љ–Є–ґ–µ–љ–Є–µ–Љ —Г—А–Њ–≤–љ—П –∞–Љ–Є–ї–Њ–Є–і–љ–Њ–≥–Њ –±–µ–ї–Ї–∞ –≤—Л—Б–Њ–Ї–Њ —Б–њ–µ—Ж–Є—Д–Є—З–љ–Њ –і–ї—П –±–Њ–ї–µ–Ј–љ–Є –Р–ї—М—Ж–≥–µ–є–Љ–µ—А–∞.
–£—Б—В–∞–љ–Њ–≤–ї–µ–љ–Њ, —З—В–Њ –Њ–і–љ–Њ–≤—А–µ–Љ–µ–љ–љ–Њ–µ —Б–љ–Є–ґ–µ–љ–Є–µ —Г—А–Њ–≤–љ—П –±–µ—В–∞-–∞–Љ–Є–ї–Њ–Є–і–∞, –њ–Њ–≤—Л—И–µ–љ–Є–µ —Г—А–Њ–≤–љ—П –∞–ї—М—Д–∞-—Б–Є–љ—Г–Ї–ї–µ–Є–љ–∞ –Є¬†—В–∞—Г-–њ—А–Њ—В–µ–Є–љ–∞, –≤¬†—В–Њ–Љ —З–Є—Б–ї–µ¬†–≥–Є–њ–µ—А—Д–Њ—Б—Д–Њ—А–Є–ї–Є—А–Њ–≤–∞–љ–љ–Њ–≥–Њ —В–∞—Г-–њ—А–Њ—В–µ–Є–љ–∞, –≤¬†—Б–њ–Є–љ–љ–Њ–Љ–Њ–Ј–≥–Њ–≤–Њ–є –ґ–Є–і–Ї–Њ—Б—В–Є –∞—Б—Б–Њ—Ж–Є–Є—А—Г–µ—В—Б—П —Б¬†–і–µ–Љ–µ–љ—Ж–Є–µ–є —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–С–Я¬†[34].
–Ь–†–Ґ¬†–≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞. –Т¬†–і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–µ –С–Я –і–Њ –љ–µ–і–∞–≤–љ–µ–≥–Њ –≤—А–µ–Љ–µ–љ–Є –Ь–†–Ґ —А–∞—Б—Б–Љ–∞—В—А–Є–≤–∞–ї–∞—Б—М –≤¬†–Њ—Б–љ–Њ–≤–љ–Њ–Љ –і–ї—П –Є—Б–Ї–ї—О—З–µ–љ–Є—П –Є–ї–Є –њ–Њ–і—В–≤–µ—А–ґ–і–µ–љ–Є—П –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є, –≤—Л–Ј—Л–≤–∞—О—Й–Є—Е –≤—В–Њ—А–Є—З–љ—Л–є –њ–∞—А–Ї–Є–љ—Б–Њ–љ–Є–Ј–Љ: –Њ–њ—Г—Е–Њ–ї–µ–є, —Б—Г–±–і—Г—А–∞–ї—М–љ—Л—Е¬†–≥–µ–Љ–∞—В–Њ–Љ, —Б–Њ—Б—Г–і–Є—Б—В–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П¬†–≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞,¬†–≥–Є–і—А–Њ—Ж–µ—Д–∞–ї–Є–Є –Є¬†—В.–і. [39, 47].
–Т¬†–њ–Њ—Б–ї–µ–і–љ–Є–µ¬†–≥–Њ–і—Л –Њ–њ–Є—Б–∞–љ—Л –љ–µ–Ї–Њ—В–Њ—А—Л–µ –љ–µ–є—А–Њ–≤–Є–Ј—Г–∞–ї–Є–Ј–∞—Ж–Є–Њ–љ–љ—Л–µ –њ—А–Є–Ј–љ–∞–Ї–Є, —Е–∞—А–∞–Ї—В–µ—А–љ—Л–µ –і–ї—П –С–Я. –Ш.–Т. –Ы–Є—В–≤–Є–љ–µ–љ–Ї–Њ –Є¬†–Ь.–Ь. –Ю–і–Є–љ–∞–Ї (2011) –њ–Њ–Ї–∞–Ј–∞–ї–Є, —З—В–Њ —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–С–Я –Є¬†–і–µ–Љ–µ–љ—Ж–Є–µ–є –љ–∞–±–ї—О–і–∞–µ—В—Б—П –∞—В—А–Њ—Д–Є—П –Ї–Њ—А—Л –≤–Є—Б–Њ—З–љ—Л—Е –Є¬†–Ј–∞—В—Л–ї–Њ—З–љ—Л—Е –і–Њ–ї–µ–є¬†–≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞. –£–Ї–∞–Ј–∞–љ–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –і–Њ—Б—В–Њ–≤–µ—А–љ–Њ –Њ—В–ї–Є—З–∞—О—В –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–С–Я –Є¬†–і–µ–Љ–µ–љ—Ж–Є–µ–є –Њ—В¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–С–Я –±–µ–Ј –і–µ–Љ–µ–љ—Ж–Є–Є –њ—А–Є —А–∞–≤–љ–Њ–є –≤—Л—А–∞–ґ–µ–љ–љ–Њ—Б—В–Є –і–≤–Є–≥–∞—В–µ–ї—М–љ—Л—Е —А–∞—Б—Б—В—А–Њ–є—Б—В–≤ [47].
–Ь–†–Ґ-–Є–Ј–Љ–µ–љ–µ–љ–Є—П, —В–Є–њ–Є—З–љ—Л–µ –і–ї—П —Б–Њ—Б—Г–і–Є—Б—В–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П¬†–≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ (–ї–∞–Ї—Г–љ—Л, –њ–µ—А–Є–≤–µ–љ—В—А–Є–Ї—Г–ї—П—А–љ—Л–є –Є¬†—Б—Г–±–Ї–Њ—А—В–Є–Ї–∞–ї—М–љ—Л–є –ї–µ–є–Ї–Њ–∞—А–µ–Њ–Ј), —З–∞—Б—В–Њ –Њ–±–љ–∞—А—Г–ґ–Є–≤–∞—О—В—Б—П —Г¬†–њ–Њ–ґ–Є–ї—Л—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†—Б–Њ—З–µ—В–∞–љ–Є–µ–Љ –С–Я, –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–є¬†–≥–Є–њ–µ—А—В–µ–љ–Ј–Є–Є, —Б–∞—Е–∞—А–љ–Њ–≥–Њ –і–Є–∞–±–µ—В–∞,¬†–≥–Є–њ–µ—А–ї–Є–њ–Є–і–µ–Љ–Є–Є,¬†–≥–Є–њ–µ—А–Ї–Њ–∞–≥—Г–ї—П—Ж–Є–Є, –љ–∞—А—Г—И–µ–љ–Є–є —Б–µ—А–і–µ—З–љ–Њ–≥–Њ —А–Є—В–Љ–∞. –°–Њ–≥–ї–∞—Б–љ–Њ –Ш.–Т. –Ы–Є—В–≤–Є–љ–µ–љ–Ї–Њ –Є¬†—Б–Њ–∞–≤—В. (2011), —Г¬†36% –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–С–Я –Є¬†–і–µ–Љ–µ–љ—Ж–Є–µ–є –љ–∞–±–ї—О–і–∞–ї—Б—П –≤—Л—А–∞–ґ–µ–љ–љ—Л–є –њ–µ—А–Є–≤–µ–љ—В—А–Є–Ї—Г–ї—П—А–љ—Л–є –ї–µ–є–Ї–Њ–∞—А–µ–Њ–Ј –≤¬†–≤–Є–і–µ ¬Ђ—И–∞–њ–Њ—З–µ–Ї¬ї —Б¬†–љ–µ—А–Њ–≤–љ—Л–Љ–Є –Ї–Њ–љ—В—Г—А–∞–Љ–Є —Г¬†–Ј–∞–і–љ–Є—Е —А–Њ–≥–Њ–≤ –±–Њ–Ї–Њ–≤—Л—Е –ґ–µ–ї—Г–і–Њ—З–Ї–Њ–≤. –°—А–µ–і–Є –±–Њ–ї—М–љ—Л—Е —Б¬†–С–Я –±–µ–Ј –і–µ–Љ–µ–љ—Ж–Є–Є –њ–Њ–і–Њ–±–љ–∞—П –ї–Њ–Ї–∞–ї–Є–Ј–∞—Ж–Є—П –ї–µ–є–Ї–Њ–∞—А–µ–Њ–Ј–∞ –±—Л–ї–∞ –Њ—В–Љ–µ—З–µ–љ–∞ –ї–Є—И—М –≤¬†6,7% —Б–ї—Г—З–∞–µ–≤. –Я—А–Є —Б–Њ—З–µ—В–∞–љ–Є–Є –С–Я —Б¬†—Б–Њ—Б—Г–і–Є—Б—В—Л–Љ –њ–Њ—А–∞–ґ–µ–љ–Є–µ–Љ¬†–≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ –і–Њ—Б—В–Њ–≤–µ—А–љ–Њ —З–∞—Й–µ –Њ–±–љ–∞—А—Г–ґ–Є–≤–∞–µ—В—Б—П –љ–∞—А—Г—И–µ–љ–Є–µ —Е–Њ–і—М–±—Л –Є¬†—А–∞–≤–љ–Њ–≤–µ—Б–Є—П. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, —Г¬†–љ–Є—Е –±–Њ–ї—М—И–µ –≤—Л—А–∞–ґ–µ–љ—Л –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л–µ —А–∞—Б—Б—В—А–Њ–є—Б—В–≤–∞ [47].
–Т–Њ–Ї—Б–µ–ї-–Њ—А–Є–µ–љ—В–Є—А–Њ–≤–∞–љ–љ–∞—П –Љ–Њ—А—Д–Њ–Љ–µ—В—А–Є—П¬†–≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ (Voxel-Based Morphometry). –Ч–∞–Ї–ї—О—З–∞–µ—В—Б—П –≤¬†–≤—Л–њ–Њ–ї–љ–µ–љ–Є–Є –≤—Л—Б–Њ–Ї–Њ–њ–Њ–ї—М–љ–Њ–є –Ь–†–Ґ –Є¬†–∞–љ–∞–ї–Є–Ј–µ –Њ–±—К–µ–Љ–∞ —А–∞–Ј–ї–Є—З–љ—Л—Е —Б—В—А—Г–Ї—В—Г—А¬†–≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞.
–Ь. Zarei (2011) –≤—Л—П–≤–Є–ї —Б–Є–ї—М–љ—Г—О –Њ—В—А–Є—Ж–∞—В–µ–ї—М–љ—Г—О –Ї–Њ—А—А–µ–ї—П—Ж–Є—О –Њ–±—К–µ–Љ–∞ —Е–≤–Њ—Б—В–∞—В–Њ–≥–Њ —П–і—А–∞ —Б¬†–Њ–±—Й–Є–Љ –±–∞–ї–ї–Њ–Љ –≤¬†—В—А–µ—В—М–µ–Љ —А–∞–Ј–і–µ–ї–µ –£–љ–Є—Д–Є—Ж–Є—А–Њ–≤–∞–љ–љ–Њ–є —А–µ–є—В–Є–љ–≥–Њ–≤–Њ–є —И–Ї–∞–ї—Л –њ—А–Є –С–Я (–≤—Л—А–∞–ґ–µ–љ–љ–Њ—Б—В—М –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л—Е –Є¬†–њ—Б–Є—Е–Є—З–µ—Б–Ї–Є—Е —А–∞—Б—Б—В—А–Њ–є—Б—В–≤) [48].
–Ф–µ–Љ–µ–љ—Ж–Є—П –њ—А–Є –С–Я –∞—Б—Б–Њ—Ж–Є–Є—А—Г–µ—В—Б—П —Б¬†—А–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–љ—Л–Љ –Є—Б—В–Њ–љ—З–µ–љ–Є–µ–Љ –Ї–Њ—А—Л¬†–≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ –≤¬†–і–Њ—А–Ј–∞–ї—М–љ—Л—Е –Є¬†–Љ–µ–і–Є–∞–ї—М–љ—Л—Е –ї–Њ–±–љ—Л—Е –Њ–±–ї–∞—Б—В—П—Е, –∞¬†—В–∞–Ї–ґ–µ –Ї–Њ—А—Л –≤–Є—Б–Њ—З–љ—Л—Е, –Ј–∞–і–љ–µ—В–µ–Љ–µ–љ–љ—Л—Е, –Ј–∞—В—Л–ї–Њ—З–љ—Л—Е –Њ–±–ї–∞—Б—В–µ–є.
–Ю–ґ–Є–≤–ї–µ–љ–љ–∞—П –і–Є—Б–Ї—Г—Б—Б–Є—П –≤–µ–і–µ—В—Б—П –њ–Њ¬†–њ–Њ–≤–Њ–і—Г —В–Є–њ–Є—З–љ—Л—Е –Ь–†–Ґ-–њ—А–Њ—П–≤–ї–µ–љ–Є–є –њ—А–Є –і–µ–Љ–µ–љ—Ж–Є–Є –љ–∞¬†—Д–Њ–љ–µ –С–Я. –Т—Л—Б–Ї–∞–Ј—Л–≤–∞—О—В—Б—П —А–∞–Ј–ї–Є—З–љ—Л–µ —В–Њ—З–Ї–Є –Ј—А–µ–љ–Є—П:
- –і–µ–Љ–µ–љ—Ж–Є—П –њ—А–Є –С–Я –∞—Б—Б–Њ—Ж–Є–Є—А—Г–µ—В—Б—П¬†–≥–ї–∞–≤–љ—Л–Љ –Њ–±—А–∞–Ј–Њ–Љ —Б¬†–Є—Б—В–Њ–љ—З–µ–љ–Є–µ–Љ –Ї–Њ—А—Л –≤–Є—Б–Њ—З–љ—Л—Е –і–Њ–ї–µ–є –Є¬†–Ј–∞–і–љ–µ–є —З–∞—Б—В–Є –њ–Њ—П—Б–љ–Њ–є –Є–Ј–≤–Є–ї–Є–љ—Л;
- –і–µ–Љ–µ–љ—Ж–Є—П –њ—А–Є –С–Я –∞—Б—Б–Њ—Ж–Є–Є—А—Г–µ—В—Б—П —Б–Њ¬†—Б–љ–Є–ґ–µ–љ–Є–µ–Љ –Њ–±—К–µ–Љ–∞ –њ—А–µ—Д—А–Њ–љ—В–∞–ї—М–љ–Њ–є, –Њ—Б—В—А–Њ–≤–Ї–Њ–≤–Њ–є, –≤–µ—А—Е–љ–µ–є –≤–Є—Б–Њ—З–љ–Њ–є –Є–Ј–≤–Є–ї–Є–љ—Л –Є¬†–њ—А–µ–Ї—Г–љ–µ—Г—Б–∞, –≤¬†—В–Њ –≤—А–µ–Љ—П –Ї–∞–Ї –њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –≤¬†–≤–Є—Б–Њ—З–љ–Њ–є –і–Њ–ї–µ –Є¬†—Б—В—А—Г–Ї—В—Г—А–∞—Е¬†–≥–Є–њ–њ–Њ–Ї–∞–Љ–њ–Њ–≤–Њ–≥–Њ –Ї—А—Г–≥–∞ –±–Њ–ї—М—И–µ —Б–≤—П–Ј–∞–љ—Л —Б¬†–±–Њ–ї–µ–Ј–љ—М—О –Р–ї—М—Ж–≥–µ–є–Љ–µ—А–∞ [49, 50].
–Я–Њ–Ј–Є—В—А–Њ–љ–љ–Њ-—Н–Љ–Є—Б—Б–Є–Њ–љ–љ–∞—П —В–Њ–Љ–Њ–≥—А–∞—Д–Є—П. –Ш—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ¬†–≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞, –њ–Њ–Ј–≤–Њ–ї—П—О—Й–µ–µ –Є–Ј–Љ–µ—А–Є—В—М –≤—Л–±—А–Њ—Б —А–∞–і–Є–Њ–∞–Ї—В–Є–≤–љ–Њ –Љ–µ—З–µ–љ–љ—Л—Е –Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Є –∞–Ї—В–Є–≤–љ—Л—Е —Е–Є–Љ–Є—З–µ—Б–Ї–Є—Е –≤–µ—Й–µ—Б—В–≤, –≤–≤–µ–і–µ–љ–љ—Л—Е –≤¬†–Ї—А–Њ–≤–µ–љ–Њ—Б–љ–Њ–µ —А—Г—Б–ї–Њ.
–Т¬†–љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П –љ–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В–Њ –Є—Б–њ–Њ–ї—М–Ј—Г–µ–Љ—Л–Љ –Є–љ–і–Є–Ї–∞—В–Њ—А–Њ–Љ –њ–Њ–Ј–Є—В—А–Њ–љ–љ–Њ-—Н–Љ–Є—Б—Б–Є–Њ–љ–љ–Њ–є —В–Њ–Љ–Њ–≥—А–∞—Д–Є–Є –Њ—Б—В–∞–µ—В—Б—П –Љ–µ—З–µ–љ–∞—П —Д–Њ—А–Љ–∞¬†–≥–ї—О–Ї–Њ–Ј—Л¬†вАУ 18F-–і–µ–Ј–Њ–Ї—Б–Є–≥–ї—О¬≠–Ї–Њ–Ј–∞ (—Д—В–Њ—А¬≠–і–µ–Ј–Њ–Ї—Б–Є–≥–ї—О–Ї–Њ–Ј–∞). –≠—В–Њ –Љ–∞—А–Ї–µ—А —Г—А–Њ–≤–љ—П –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ–∞¬†–≥–ї—О–Ї–Њ–Ј—Л –≤¬†–Љ–Њ–Ј–≥–µ, –њ–Њ–Ї–∞–Ј—Л–≤–∞—О—Й–Є–є —А–∞—Б–њ—А–µ–і–µ–ї–µ–љ–Є–µ –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –Ї–ї–µ—В–Њ–Ї. –Р–Ї—В–Є–≤–љ–Њ –њ—А–Є–Љ–µ–љ—П–µ—В—Б—П –Є¬†–Я–Є—В—В—Б–±—Г—А–≥—Б–Ї–∞—П —Б—Г–±—Б—В–∞–љ—Ж–Є—П B¬†вАУ –Љ–∞—А–Ї–µ—А –Њ—В–ї–Њ–ґ–µ–љ–Є—П –±–µ—В–∞-–∞–Љ–Є–ї–Њ–Є–і–∞ –≤¬†–≥–Њ–ї–Њ–≤–љ–Њ–Љ –Љ–Њ–Ј–≥–µ.
–°–Њ–≥–ї–∞—Б–љ–Њ –њ—А–Њ–≤–µ–і–µ–љ–љ—Л–Љ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П–Љ, —Г¬†–±–Њ–ї—М–љ—Л—Е –С–Я –љ–∞¬†—А–∞–љ–љ–Є—Е —Б—В–∞–і–Є—П—Е –±—Л–ї –≤—Л—П–≤–ї–µ–љ –ї–Є—И—М –љ–µ–Ј–љ–∞—З–Є—В–µ–ї—М–љ—Л–є¬†–≥–Є–њ–Њ–Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ¬†–≥–ї—О–Ї–Њ–Ј—Л –≤¬†—А–∞–Ј–ї–Є—З–љ—Л—Е –Њ—В–і–µ–ї–∞—Е –Ї–Њ—А—Л¬†–≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞. –Э–∞¬†2,5вАУ3-–є¬†—Б—В–∞–і–Є–Є –С–Я –±–µ–Ј –і–µ–Љ–µ–љ—Ж–Є–Є –Њ—В–Љ–µ—З–∞–ї—Б—П¬†–≥–Є–њ–Њ–Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ –≤¬†—Е–≤–Њ—Б—В–∞—В—Л—Е —П–і—А–∞—Е –Є¬†–і–Њ—А—Б–Њ–ї–∞—В–µ—А–∞–ї—М–љ–Њ–є –њ—А–µ—Д—А–Њ–љ—В–∞–ї—М–љ–Њ–є –Ї–Њ—А–µ –њ—А–Є —Б–Њ—Е—А–∞–љ–љ–Њ–Љ –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ–µ –≤¬†–і—А—Г–≥–Є—Е —Б—В—А—Г–Ї—В—Г—А–∞—Е –Љ–Њ–Ј–≥–∞ [49, 51]. –£–Ї–∞–Ј–∞–љ–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –Ї–Њ—А—А–µ–ї–Є—А–Њ–≤–∞–ї–Є —Б¬†—А–µ–Ј—Г–ї—М—В–∞—В–∞–Љ–Є –љ–µ–є—А–Њ–њ—Б–Є—Е–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е —В–µ—Б—В–Њ–≤, —Б–Њ–≥–ї–∞—Б–љ–Њ –Ї–Њ—В–Њ—А—Л–Љ —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ –Є–Љ–µ–ї–Є –Љ–µ—Б—В–Њ —Б–љ–Є–ґ–µ–љ–Є–µ —Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В–Є –Ї¬†–Њ–±–Њ–±—Й–µ–љ–Є—О, –∞–љ–∞–ї–Є–Ј—Г –Є¬†—Б–Є–љ—В–µ–Ј—Г –Є–љ—Д–Њ—А–Љ–∞—Ж–Є–Є, –љ–µ–≥—А—Г–±—Л–µ –љ–∞—А—Г—И–µ–љ–Є—П –њ–∞–Љ—П—В–Є –Є¬†–≤–љ–Є–Љ–∞–љ–Є—П, –Ј—А–Є—В–µ–ї—М–љ–Њ-–њ—А–Њ—Б—В—А–∞–љ—Б—В–≤–µ–љ–љ—Л—Е —Д—Г–љ–Ї—Ж–Є–є. –Т¬†–≥—А—Г–њ–њ–µ –±–Њ–ї—М–љ—Л—Е –С–Я —Б¬†–і–µ–Љ–µ–љ—Ж–Є–µ–є –љ–∞–±–ї—О–і–∞–ї–Њ—Б—М –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ–µ –і–≤—Г—Б—В–Њ—А–Њ–љ–љ–µ–µ —Б–љ–Є–ґ–µ–љ–Є–µ –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ–∞ –Ї–Њ—А—Л –±–Њ–ї—М—И–Є—Е –њ–Њ–ї—Г—И–∞—А–Є–є¬†–≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞. –£¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–С–Я, –і–µ–Љ–µ–љ—Ж–Є–µ–є –Є¬†–Ј—А–Є—В–µ–ї—М–љ—Л–Љ–Є¬†–≥–∞–ї–ї—О—Ж–Є–љ–∞—Ж–Є—П–Љ–Є –љ–∞–Є–±–Њ–ї—М—И–Є–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –Њ–±–љ–∞—А—Г–ґ–µ–љ—Л –≤¬†–Ј–∞—В—Л–ї–Њ—З–љ–Њ–є –Ї–Њ—А–µ, –њ–Њ—П—Б–љ—Л—Е –Є–Ј–≤–Є–ї–Є–љ–∞—Е, –Њ—А–±–Є—В–Њ—Д—А–Њ–љ—В–∞–ї—М–љ–Њ–є –Ї–Њ—А–µ –Є¬†—Б—В—А—Г–Ї—В—Г—А–∞—Е¬†–≥–Є–њ–њ–Њ–Ї–∞–Љ–њ–Њ–≤–Њ–≥–Њ –Ї—А—Г–≥–∞ [51].
–Ы–µ—З–µ–љ–Є–µ
–Э–µ—Д–∞—А–Љ–∞–Ї–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –Љ–µ—В–Њ–і—Л
–Ъ¬†–Њ–±—Й–Є–Љ –Љ–µ—А–∞–Љ –Њ—В–љ–Њ—Б—П—В—Б—П –Є–љ—Д–Њ—А–Љ–∞—Ж–Є–Њ–љ–љ—Л–µ –±–µ—Б–µ–і—Л —Б¬†–њ–∞—Ж–Є–µ–љ—В–Њ–Љ –Є¬†–ї–Є—Ж–∞–Љ–Є, —Г—З–∞—Б—В–≤—Г—О—Й–Є–Љ–Є –≤¬†—Г—Е–Њ–і–µ –Ј–∞ –љ–Є–Љ, —А–µ–Ї–Њ–Љ–µ–љ–і–∞—Ж–Є–Є –њ–Њ¬†—Б–Њ—Е—А–∞–љ–µ–љ–Є—О –і–Њ—Б—В–∞—В–Њ—З–љ–Њ–є –њ—Б–Є—Е–Є—З–µ—Б–Ї–Њ–є –Є¬†—Д–Є–Ј–Є—З–µ—Б–Ї–Њ–є –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є, –Є—Б–Ї–ї—О—З–µ–љ–Є–µ –њ—Б–Є—Е–Њ—Н–Љ–Њ—Ж–Є–Њ–љ–∞–ї—М–љ—Л—Е –њ–µ—А–µ–≥—А—Г–Ј–Њ–Ї, –љ–µ–±–ї–∞–≥–Њ–њ—А–Є—П—В–љ—Л—Е –≤–Њ–Ј–і–µ–є—Б—В–≤–Є–є –Њ–Ї—А—Г–ґ–∞—О—Й–µ–є —Б—А–µ–і—Л.
–Я–Њ–ї–Њ–ґ–Є—В–µ–ї—М–љ–Њ –≤–ї–Є—П—О—В –љ–∞¬†–Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л–µ —Д—Г–љ–Ї—Ж–Є–Є —Д–Є–Ј–Є—З–µ—Б–Ї–∞—П –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –Є¬†—В—А–µ–љ–Є—А–Њ–≤–Ї–Є: –∞—Н—А–Њ–±–љ—Л–µ –љ–∞–≥—А—Г–Ј–Ї–Є, —Г–њ—А–∞–ґ–љ–µ–љ–Є—П –љ–∞¬†—Б–Њ–њ—А–Њ—В–Є–≤–ї–µ–љ–Є–µ, —А–∞—Б—В—П–ґ–µ–љ–Є–µ, —Б–Є–ї–Њ–≤–Њ–є –Є¬†–±–∞–ї–∞–љ—Б-—В—А–µ–љ–Є–љ–≥. –Я—А–µ–і–њ–Њ—З—В–µ–љ–Є–µ –Њ—В–і–∞–µ—В—Б—П –∞—Н—А–Њ–±–љ–Њ–Љ—Г —В—А–µ–љ–Є–љ–≥—Г, –њ–Њ—Б–Ї–Њ–ї—М–Ї—Г –њ—А–µ–і–њ–Њ–ї–∞–≥–∞–µ—В—Б—П, —З—В–Њ –Њ–љ —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г–µ—В —Г–ї—Г—З—И–µ–љ–Є—О –Ї–∞–Ї —Д—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ–Њ–є —Ж–µ—А–µ–±—А–∞–ї—М–љ–Њ–є –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є, —В–∞–Ї –Є,¬†–≤–Њ–Ј–Љ–Њ–ґ–љ–Њ, —Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–Є—О –љ–Њ–≤—Л—Е –Љ–µ–ґ—Б–Є–љ–∞–њ—В–Є—З–µ—Б–Ї–Є—Е –≤–Ј–∞–Є–Љ–Њ–і–µ–є—Б—В–≤–Є–є –Є¬†—Б–љ–Є–ґ–∞–µ—В —Б—В–µ–њ–µ–љ—М —Ж–µ—А–µ–±—А–∞–ї—М–љ–Њ–є¬†–≥–Є–њ–Њ–њ–µ—А—Д—Г–Ј–Є–Є –Є¬†–≥–Є–њ–Њ–Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ–∞ [46].
–Ъ–Њ–≥–љ–Є—В–Є–≤–љ—Л–є —В—А–µ–љ–Є–љ–≥ —Г–ї—Г—З—И–∞–µ—В –≤–љ–Є–Љ–∞–љ–Є–µ, –Є—Б–њ–Њ–ї–љ–Є—В–µ–ї—М–љ—Л–µ —Д—Г–љ–Ї—Ж–Є–Є, –њ–∞–Љ—П—В—М –Є¬†–Ј—А–Є—В–µ–ї—М–љ–Њ-–њ—А–Њ—Б—В—А–∞–љ—Б—В–≤–µ–љ–љ—Л–µ —Д—Г–љ–Ї—Ж–Є–Є —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–С–Я –Є¬†–і–µ–Љ–µ–љ—Ж–Є–µ–є [52]. –Я—А–Њ–≤–µ–і–µ–љ–Њ —И–µ—Б—В—М —А–∞–љ–і–Њ–Љ–Є–Ј–Є—А–Њ–≤–∞–љ–љ—Л—Е –Ї–Њ–љ—В—А–Њ–ї–Є—А—Г–µ–Љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –њ–Њ¬†—Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В–Є –і–∞–љ–љ–Њ–≥–Њ –Љ–µ—В–Њ–і–∞ –ї–µ—З–µ–љ–Є—П, –њ—П—В—М –Є–Ј¬†–Ї–Њ—В–Њ—А—Л—Е –њ–Њ–Ї–∞–Ј–∞–ї–Є –њ–Њ–ї–Њ–ґ–Є—В–µ–ї—М–љ—Л–є —А–µ–Ј—Г–ї—М—В–∞—В, –≤¬†–Њ–і–љ–Њ–Љ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є –њ–Њ–ї—Г—З–µ–љ—Л –і–Њ—Б—В–Њ–≤–µ—А–љ—Л–µ –і–Њ–Ї–∞–Ј–∞—В–µ–ї—М—Б—В–≤–∞ —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В–Є –Љ–µ—В–Њ–і–∞.
–Ъ–Њ–≥–љ–Є—В–Є–≤–љ—Л–є —В—А–µ–љ–Є–љ–≥ –њ—А–µ–і—Б—В–∞–≤–ї—П–µ—В —Б–Њ–±–Њ–є —Б–њ–µ—Ж–Є–∞–ї—М–љ—Л–µ –њ—А–Њ–≥—А–∞–Љ–Љ—Л –Є¬†–Љ–µ—В–Њ–і–Є–Ї–Є –і–ї—П —В—А–µ–љ–Є—А–Њ–≤–Ї–Є –њ–∞–Љ—П—В–Є, –≤–љ–Є–Љ–∞–љ–Є—П –Є¬†–і—А—Г–≥–Є—Е –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л—Е —Д—Г–љ–Ї—Ж–Є–є, –љ–∞–њ—А–∞–≤–ї–µ–љ–љ—Л–µ –љ–∞¬†–њ–Њ–і–і–µ—А–ґ–∞–љ–Є–µ –Њ–њ—В–Є–Љ–∞–ї—М–љ–Њ–≥–Њ –Є–љ—В–µ–ї–ї–µ–Ї—В—Г–∞–ї—М–љ–Њ–≥–Њ —Г—А–Њ–≤–љ—П, —А–∞–Ј–≤–Є—В–Є–µ —Б–љ–Є–ґ–µ–љ–љ—Л—Е –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л—Е —Д—Г–љ–Ї—Ж–Є–є, –∞¬†—В–∞–Ї–ґ–µ –љ–∞¬†–Њ–±—Г—З–µ–љ–Є–µ —Б—В—А–∞—В–µ–≥–Є—П–Љ –Ї–Њ–Љ–њ–µ–љ—Б–∞—Ж–Є–Є. –Т—Л–і–µ–ї—П—О—В –і–≤–∞ —В–Є–њ–∞ –Ї–Њ–≥–љ–Є—В–Є–≤–љ–Њ–≥–Њ —В—А–µ–љ–Є–љ–≥–∞: –Ї–Њ–Љ–њ–µ–љ—Б–∞—В–Њ—А–љ—Л–є –Є¬†–≤–Њ—Б—Б—В–∞–љ–Њ–≤–Є—В–µ–ї—М–љ—Л–є. –Т¬†—Е–Њ–і–µ –Ї–Њ–Љ–њ–µ–љ—Б–∞—В–Њ—А–љ–Њ–≥–Њ –Ї–Њ–≥–љ–Є—В–Є–≤–љ–Њ–≥–Њ —В—А–µ–љ–Є–љ–≥–∞ –њ–∞—Ж–Є–µ–љ—В –Њ–±—Г—З–∞–µ—В—Б—П –љ–Њ–≤—Л–Љ —Б—В—А–∞—В–µ–≥–Є—П–Љ —А–µ—И–µ–љ–Є—П –њ–Њ—Б—В–∞–≤–ї–µ–љ–љ–Њ–є –Ј–∞–і–∞—З–Є —З–µ—А–µ–Ј —Б–Њ—Е—А–∞–љ–љ—Л–µ –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л–µ —Д—Г–љ–Ї—Ж–Є–Є. –Я—А–Є –≤–Њ—Б—Б—В–∞–љ–Њ–≤–Є—В–µ–ї—М–љ–Њ–Љ –Ї–Њ–≥–љ–Є—В–Є–≤–љ–Њ–Љ —В—А–µ–љ–Є–љ–≥–µ –Љ–µ—А–Њ–њ—А–Є—П—В–Є—П –љ–∞—Ж–µ–ї–µ–љ—Л –љ–∞¬†—Г–ї—Г—З—И–µ–љ–Є–µ –њ–Њ–≤—А–µ–ґ–і–µ–љ–љ—Л—Е –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л—Е —Д—Г–љ–Ї—Ж–Є–є [46].
–Ь–µ–і–Є–Ї–∞–Љ–µ–љ—В–Њ–Ј–љ–∞—П —В–µ—А–∞–њ–Є—П
–Ф–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ –њ—А–Є –С–Я —Б¬†–і–µ–Љ–µ–љ—Ж–Є–µ–є –Є–Љ–µ–µ—В –Љ–µ—Б—В–Њ —Е–Њ–ї–Є–љ–µ—А–≥–Є—З–µ—Б–Ї–Є–є –і–µ—Д–Є—Ж–Є—В, –≤¬†—Б–≤—П–Ј–Є —Б¬†—З–µ–Љ –љ–∞–Є–±–Њ–ї–µ–µ –Њ–±–Њ—Б–љ–Њ–≤–∞–љ–љ–Њ –њ—А–Є–Љ–µ–љ–µ–љ–Є–µ –Є–љ–≥–Є–±–Є—В–Њ—А–Њ–≤ –∞—Ж–µ—В–Є–ї—Е–Њ–ї–Є–љ—Н—Б—В–µ—А–∞–Ј—Л. –Ъ–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –њ–Њ–Ї–∞–Ј–∞–ї–Є, —З—В–Њ —В–∞–Ї–Є–Љ –њ–∞—Ж–Є–µ–љ—В–∞–Љ –Љ–Њ–≥—Г—В –љ–∞–Ј–љ–∞—З–∞—В—М—Б—П –≤—Б–µ –Є–љ–≥–Є–±–Є—В–Њ—А—Л –∞—Ж–µ—В–Є–ї—Е–Њ–ї–Є–љ—Н—Б—В–µ—А–∞–Ј—Л, –Ј–∞—А–µ–≥–Є—Б—В—А–Є—А–Њ–≤–∞–љ–љ—Л–µ –љ–∞¬†–љ–∞—Б—В–Њ—П—Й–Є–є –Љ–Њ–Љ–µ–љ—В: –і–Њ–љ–µ–њ–µ–Ј–Є–ї, —А–Є–≤–∞—Б—В–Є–≥–Љ–Є–љ,¬†–≥–∞–ї–∞–љ—В–∞–Љ–Є–љ.
–°–Њ–≥–ї–∞—Б–љ–Њ –Љ–µ—В–∞–∞–љ–∞–ї–Є–Ј—Г –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В–Є –Є–љ–≥–Є–±–Є—В–Њ—А–Њ–≤ –∞—Ж–µ—В–Є–ї—Е–Њ–ї–Є–љ—Н—Б—В–µ—А–∞–Ј—Л –њ—А–Є –С–Я —Б –і–µ–Љ–µ–љ—Ж–Є–µ–є, –љ–∞¬†—Д–Њ–љ–µ –ї–µ—З–µ–љ–Є—П —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Г–Љ–µ–љ—М—И–∞–µ—В—Б—П –≤—Л—А–∞–ґ–µ–љ–љ–Њ—Б—В—М –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л—Е –љ–∞—А—Г—И–µ–љ–Є–є, —Г–ї—Г—З—И–∞—О—В—Б—П –њ–Њ–≤–µ–і–µ–љ–Є–µ –Є¬†–Њ–±—Й–µ–µ –Ї–∞—З–µ—Б—В–≤–Њ –ґ–Є–Ј–љ–Є [53]. –Э–∞–Ј–љ–∞—З–µ–љ–Є–µ –Є–љ–≥–Є–±–Є—В–Њ—А–Њ–≤ –∞—Ж–µ—В–Є–ї—Е–Њ–ї–Є–љ—Н—Б—В–µ—А–∞–Ј—Л —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г–µ—В —Г–Љ–µ–љ—М—И–µ–љ–Є—О –≤—Л—А–∞–ґ–µ–љ–љ–Њ—Б—В–Є –њ—Б–Є—Е–Є—З–µ—Б–Ї–Є—Е —А–∞—Б—Б—В—А–Њ–є—Б—В–≤, —В–∞–Ї–Є—Е –Ї–∞–Ї –∞–њ–∞—В–Є—П, —В—А–µ–≤–Њ–ґ–љ–Њ—Б—В—М,¬†–≥–∞–ї–ї—О—Ж–Є–љ–∞—Ж–Є–Є –Є¬†–і–µ–ї–Є—А–Є–є [54]. –Э–∞¬†—Д–Њ–љ–µ –ї–µ—З–µ–љ–Є—П –≤—Л—А–∞–ґ–µ–љ–љ–Њ—Б—В—М –і–≤–Є–≥–∞—В–µ–ї—М–љ—Л—Е —А–∞—Б—Б—В—А–Њ–є—Б—В–≤ –љ–µ¬†–љ–∞—А–∞—Б—В–∞–µ—В.
–Т—В–Њ—А–Њ–є –Ї–ї–∞—Б—Б –њ—А–µ–њ–∞—А–∞—В–Њ–≤, –Є—Б–њ–Њ–ї—М–Ј—Г—О—Й–Є–є—Б—П –њ—А–Є –С–Я —Б¬†–і–µ–Љ–µ–љ—Ж–Є–µ–є,¬†вАУ –∞–љ—В–∞–≥–Њ–љ–Є—Б—В—Л —А–µ—Ж–µ–њ—В–Њ—А–Њ–≤ –Ї¬†N-–Љ–µ—В–Є–ї-D-–∞—Б–њ–∞—А—В–∞—В—Г. –Я—А–µ–і—Б—В–∞–≤–Є—В–µ–ї—М —Н—В–Њ–≥–Њ –Ї–ї–∞—Б—Б–∞¬†вАУ –Љ–µ–Љ–∞–љ—В–Є–љ. –Я—А–Њ–≤–µ–і–µ–љ–љ—Л–µ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –њ–Њ–Ї–∞–Ј–∞–ї–Є, —З—В–Њ –њ—А–µ–њ–∞—А–∞—В —Н—Д—Д–µ–Ї—В–Є–≤–µ–љ –љ–∞¬†—Б—В–∞–і–Є–Є —Г–Љ–µ—А–µ–љ–љ—Л—Е –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л—Е —А–∞—Б—Б—В—А–Њ–є—Б—В–≤ –Є¬†—Г –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†—Б–Њ—З–µ—В–∞–љ–Є–µ–Љ –С–Я –Є¬†–і–µ–Љ–µ–љ—Ж–Є–Є, –Є¬†—Г –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–С–Я –Є¬†–Њ—В—Б—Г—В—Б—В–≤–Є–µ–Љ –і–µ–Љ–µ–љ—Ж–Є–Є. –Э–∞¬†—Д–Њ–љ–µ –њ—А–Є–µ–Љ–∞ –Љ–µ–Љ–∞–љ—В–Є–љ–∞ –і–Њ—Б—В–Њ–≤–µ—А–љ–Њ —Б–љ–Є–ґ–∞–µ—В—Б—П –≤—Л—А–∞–ґ–µ–љ–љ–Њ—Б—В—М –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л—Е –љ–∞—А—Г—И–µ–љ–Є–є, –∞–њ–∞—В–Є–Є, –њ–Њ–≤–µ–і–µ–љ—З–µ—Б–Ї–Є—Е —А–∞—Б—Б—В—А–Њ–є—Б—В–≤. –Т—Л—А–∞–ґ–µ–љ–љ–Њ—Б—В—М –Ј—А–Є—В–µ–ї—М–љ—Л—Е¬†–≥–∞–ї–ї—О—Ж–Є–љ–∞—Ж–Є–є, –Њ–і–љ–∞–Ї–Њ, –Њ–±—Л—З–љ–Њ –Њ—Б—В–∞–µ—В—Б—П –љ–µ–Є–Ј–Љ–µ–љ–љ–Њ–є –Є–ї–Є —Г–Љ–µ–љ—М—И–∞–µ—В—Б—П –љ–µ–Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ [52].
–£¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–С–Я –±–µ–Ј –і–µ–Љ–µ–љ—Ж–Є–Є –Љ–Њ–ґ–љ–Њ –Њ–ґ–Є–і–∞—В—М –њ–Њ–ї–Њ–ґ–Є—В–µ–ї—М–љ—Л–є —Н—Д—Д–µ–Ї—В –Њ—В¬†–њ—А–Є–µ–Љ–∞ –њ—А–µ–њ–∞—А–∞—В–Њ–≤ –ї–µ–≤–Њ–і–Њ–њ—Л –≤¬†—В–Њ–Љ —З–Є—Б–ї–µ –≤¬†–Њ—В–љ–Њ—И–µ–љ–Є–Є –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л—Е —Д—Г–љ–Ї—Ж–Є–є. –Ґ–∞–Ї, —Б–Њ–≥–ї–∞—Б–љ–Њ –њ–Њ–ї—Г—З–µ–љ–љ—Л–Љ –і–∞–љ–љ—Л–Љ, –љ–∞¬†—Д–Њ–љ–µ –Ї–Њ—А—А–µ–Ї—Ж–Є–Є –і–Њ–Ј—Л –ї–µ–≤–Њ–і–Њ–њ—Л —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–С–Я –±–µ–Ј –і–µ–Љ–µ–љ—Ж–Є–Є —Г—Б–Ї–Њ—А—П–ї–Є—Б—М –њ—Б–Є—Е–Є—З–µ—Б–Ї–Є–µ —А–µ–∞–Ї—Ж–Є–Є, —Б–љ–Є–ґ–∞–ї–∞—Б—М –Є–љ–µ—А—В–љ–Њ—Б—В—М –Љ—Л—И–ї–µ–љ–Є—П, –Њ–±–ї–µ–≥—З–∞–ї—Б—П –њ–µ—А–µ—Е–Њ–і —Б¬†–Њ–і–љ–Њ–≥–Њ —Н—В–∞–њ–∞ –Ј–∞–і–∞—З–Є –љ–∞¬†–і—А—Г–≥–Њ–є. –Ю–і–љ–∞–Ї–Њ —А–µ–Ј—Г–ї—М—В–∞—В—Л –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В–Є –њ—А–µ–њ–∞—А–∞—В–Њ–≤ –ї–µ–≤–Њ–і–Њ–њ—Л —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†—Б–Њ—З–µ—В–∞–љ–Є–µ–Љ –С–Я –Є¬†–і–µ–Љ–µ–љ—Ж–Є–Є —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г—О—В, —З—В–Њ –љ–Є–Ї–∞–Ї–Њ–≥–Њ –≤–ї–Є—П–љ–Є—П –љ–∞¬†–Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л–µ —Д—Г–љ–Ї—Ж–Є–Є —Г¬†—Н—В–Є—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –љ–∞–Ј–љ–∞—З–µ–љ–Є–µ –Є–ї–Є –Є–Ј–Љ–µ–љ–µ–љ–Є–µ –і–Њ–Ј—Л –њ—А–µ–њ–∞—А–∞—В–Њ–≤ –ї–µ–≤–Њ–і–Њ–њ—Л —Г–ґ–µ –љ–µ¬†–Њ–Ї–∞–Ј—Л–≤–∞–µ—В [55].
–Т –љ–∞—Б—В–Њ—П—Й–µ–µ –≤—А–µ–Љ—П –њ—А–Њ—Е–Њ–і–Є—В –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ –Є—Б–њ—Л—В–∞–љ–Є—П –љ–Є–ї–Њ—В–Є–љ–Є–±, —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г—О—Й–Є–є –і–µ–≥—А–∞–і–∞—Ж–Є–Є –∞–ї—М—Д–∞-—Б–Є–љ—Г–Ї–ї–µ–Є–љ–∞ —Б¬†–њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–є –Ї–Њ–љ—Д–Њ—А–Љ–∞—Ж–Є–µ–є (–њ—А–µ–њ–∞—А–∞—В –Ј–∞—А–µ–≥–Є—Б—В—А–Є—А–Њ–≤–∞–љ –і–ї—П –ї–µ—З–µ–љ–Є—П —Е—А–Њ–љ–Є—З–µ—Б–Ї–Њ–≥–Њ –Љ–Є–µ–ї–Њ–ї–µ–є–Ї–Њ–Ј–∞, –Њ–±–ї–∞–і–∞–µ—В —Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В—М—О –Є–љ–≥–Є–±–Є—А–Њ–≤–∞—В—М —В–Є—А–Њ–Ј–Є–љ–Ї–Є–љ–∞–Ј—Г –Ї–ї–∞—Б—В–µ—А–љ–Њ–≥–Њ —А–µ–≥–Є–Њ–љ–∞ —В–Њ—З–µ—З–љ–Њ–≥–Њ —А–∞–Ј—А—Л–≤–∞ –Р–±–µ–ї—М—Б–Њ–љ–∞)¬†[56].
–Ш–љ—В–µ–љ—Б–Є–≤–љ–Њ —А–∞–Ј—А–∞–±–∞—В—Л–≤–∞–µ—В—Б—П –Є–Љ–Љ—Г–љ–Њ—В–µ—А–∞–њ–Є—П¬†вАУ —Б–Њ–Ј–і–∞–љ–Є–µ –∞–љ—В–Є—В–µ–ї –њ—А–Њ—В–Є–≤ –∞–ї—М—Д–∞-—Б–Є–љ—Г–Ї–ї–µ–Є–љ–∞. –Э–µ—Б–Ї–Њ–ї—М–Ї–Њ –∞–љ—В–Є—В–µ–ї –њ—А–Њ–і–µ–Љ–Њ–љ—Б—В—А–Є—А–Њ–≤–∞–ї–Є –∞–Ї—В–Є–≤–љ–Њ—Б—В—М in¬†vitro –Є¬†–љ–∞ –ґ–Є–≤–Њ—В–љ—Л—Е –Љ–Њ–і–µ–ї—П—Е. –Э–µ —В–∞–Ї –і–∞–≤–љ–Њ –±—Л–ї–Є –Њ–±–љ–∞—А–Њ–і–Њ–≤–∞–љ—Л —А–µ–Ј—Г–ї—М—В–∞—В—Л –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В–Є –Љ–Њ–љ–Њ–Ї–ї–Њ–љ–∞–ї—М–љ—Л—Е –∞–љ—В–Є—В–µ–ї PRX002 –Ї¬†–∞–ї—М—Д–∞-—Б–Є–љ—Г–Ї–ї–µ–Є–љ—Г —Г¬†40 –Ј–і–Њ—А–Њ–≤—Л—Е –і–Њ–±—А–Њ–≤–Њ–ї—М—Ж–µ–≤ [56]. –С–µ–Ј—Г—Б–ї–Њ–≤–љ–Њ, —Б–µ–є—З–∞—Б –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –љ–∞—Е–Њ–і—П—В—Б—П –љ–∞ –љ–∞—З–∞–ї—М–љ–Њ–Љ —Н—В–∞–њ–µ, —В—А–µ–±—Г–µ—В—Б—П –і–∞–ї—М–љ–µ–є—И–µ–µ –Є–Ј—Г—З–µ–љ–Є–µ –Ї–∞–Ї –±–µ–Ј–Њ–њ–∞—Б–љ–Њ—Б—В–Є, —В–∞–Ї –Є —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В–Є —Н—В–Њ–≥–Њ –Љ–µ—В–Њ–і–∞ –ї–µ—З–µ–љ–Є—П.
–Ъ—Г–њ–Є—А–Њ–≤–∞–љ–Є–µ –љ–µ–є—А–Њ–њ—Б–Є—Е–Є–∞—В—А–Є—З–µ—Б–Ї–Є—Е¬†—Б–Є–Љ–њ—В–Њ–Љ–Њ–≤
–Я—Б–Є—Е–Є–∞—В—А–Є—З–µ—Б–Ї–Є–µ –Є¬†–њ–Њ–≤–µ–і–µ–љ—З–µ—Б–Ї–Є–µ —Б–Є–Љ–њ—В–Њ–Љ—Л —З–∞—Б—В–Њ –і–µ–Ј–∞–і–∞–њ—В–Є—А—Г—О—В –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –Є¬†–Є—Е —А–Њ–і—Б—В–≤–µ–љ–љ–Є–Ї–Њ–≤ –±–Њ–ї—М—И–µ, —З–µ–Љ –і–≤–Є–≥–∞—В–µ–ї—М–љ—Л–µ –њ—А–Њ—П–≤–ї–µ–љ–Є—П. –Ґ–µ—А–∞–њ–Є—П –Є–љ–≥–Є–±–Є—В–Њ—А–∞–Љ–Є –∞—Ж–µ—В–Є–ї—Е–Њ–ї–Є–љ—Н—Б—В–µ—А–∞–Ј—Л –Љ–Њ–ґ–µ—В –Є–Љ–µ—В—М –њ–Њ–ї–Њ–ґ–Є—В–µ–ї—М–љ—Л–є —Н—Д—Д–µ–Ї—В –≤¬†–Њ—В–љ–Њ—И–µ–љ–Є–Є —А—П–і–∞ —Б–Є–Љ–њ—В–Њ–Љ–Њ–≤: –Ј—А–Є—В–µ–ї—М–љ—Л—Е¬†–≥–∞–ї–ї—О—Ж–Є–љ–∞—Ж–Є–є, –љ–∞—А—Г—И–µ–љ–Є—П —Ж–Є–Ї–ї–∞ ¬Ђ—Б–Њ–љ¬†вАУ –±–Њ–і—А—Б—В–≤–Њ–≤–∞–љ–Є–µ¬ї, –∞–њ–∞—В–Є–Є, –±—А–µ–і–Њ–≤—Л—Е —А–∞—Б—Б—В—А–Њ–є—Б—В–≤ [54]. –Т¬†–Љ–µ–љ—М—И–µ–є —Б—В–µ–њ–µ–љ–Є –Њ–љ–∞ –≤–ї–Є—П–µ—В –љ–∞¬†–≤—Л—А–∞–ґ–µ–љ–љ–Њ—Б—В—М –∞–≥—А–µ—Б—Б–Є–Є –Є¬†–њ—Б–Є—Е–Њ–Љ–Њ—В–Њ—А–љ–Њ–≥–Њ –≤–Њ–Ј–±—Г–ґ–і–µ–љ–Є—П, –≤¬†—В–∞–Ї–Є—Е —Б–ї—Г—З–∞—П—Е –њ—А–µ–њ–∞—А–∞—В–∞–Љ–Є –≤—Л–±–Њ—А–∞ —П–≤–ї—П—О—В—Б—П –∞–љ—В–∞–≥–Њ–љ–Є—Б—В—Л D2-—А–µ—Ж–µ–њ—В–Њ—А–Њ–≤. –Э–µ–Њ–±—Е–Њ–і–Є–Љ–Њ –Њ—В–Љ–µ—В–Є—В—М, —З—В–Њ –њ—А–Є–Љ–µ–љ–µ–љ–Є–µ —В–Є–њ–Є—З–љ—Л—Е –љ–µ–є—А–Њ–ї–µ–њ—В–Є–Ї–Њ–≤ –њ—А–Є –С–Я —Б¬†–і–µ–Љ–µ–љ—Ж–Є–µ–є –∞–±—Б–Њ–ї—О—В–љ–Њ –љ–µ–і–Њ–њ—Г—Б—В–Є–Љ–Њ –Є–Ј-–Ј–∞ –≤—Л—Б–Њ–Ї–Њ–є –≤–µ—А–Њ—П—В–љ–Њ—Б—В–Є –љ–∞—А–∞—Б—В–∞–љ–Є—П –≤—Л—А–∞–ґ–µ–љ–љ–Њ—Б—В–Є –і–≤–Є–≥–∞—В–µ–ї—М–љ—Л—Е —А–∞—Б—Б—В—А–Њ–є—Б—В–≤ –Є¬†—А–∞–Ј–≤–Є—В–Є—П –љ–µ–є—А–Њ–ї–µ–њ—В–Є—З–µ—Б–Ї–Њ–≥–Њ —Б–Є–љ–і—А–Њ–Љ–∞ [57]. –Т–Њ–Ј–Љ–Њ–ґ–µ–љ –њ—А–Є–µ–Љ —В–Њ–ї—М–Ї–Њ –∞—В–Є–њ–Є—З–љ—Л—Е –љ–µ–є—А–Њ–ї–µ–њ—В–Є–Ї–Њ–≤ –Є¬†–Њ–±—П–Ј–∞—В–µ–ї—М–љ–Њ –њ–Њ–і –Ї–Њ–љ—В—А–Њ–ї–µ–Љ –≤—А–∞—З–∞. –Э–∞–Ј–љ–∞—З–∞—В—М –њ—А–µ–њ–∞—А–∞—В—Л –Є¬†–љ–∞—А–∞—Й–Є–≤–∞—В—М –і–Њ–Ј—Г —Ж–µ–ї–µ—Б–Њ–Њ–±—А–∞–Ј–љ–Њ –≤¬†—Г—Б–ї–Њ–≤–Є—П—Е —Б—В–∞—Ж–Є–Њ–љ–∞—А–∞.
–Э–∞–Є–±–Њ–ї—М—И–Є–µ –і–Њ–Ї–∞–Ј–∞—В–µ–ї—М—Б—В–≤–∞ —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В–Є, —Б–Њ–≥–ї–∞—Б–љ–Њ —А–µ–Ј—Г–ї—М—В–∞—В–∞–Љ –њ—А–Њ–≤–µ–і–µ–љ–љ—Л—Е –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є, –і–Њ—Б—В–Є–≥–љ—Г—В—Л –і–ї—П –Ї–ї–Њ–Ј–∞–њ–Є–љ–∞. –Я—А–µ–њ–∞—А–∞—В –љ–µ¬†–Њ–Ї–∞–Ј—Л–≤–∞–µ—В –Ј–љ–∞—З–Є–Љ–Њ–≥–Њ –≤–ї–Є—П–љ–Є—П –љ–∞¬†–Љ–Њ—В–Њ—А–љ—Л–µ —Д—Г–љ–Ї—Ж–Є–Є. –Я—А–Є–µ–Љ –Ї–ї–Њ–Ј–∞–њ–Є–љ–∞ —В—А–µ–±—Г–µ—В –µ–ґ–µ–љ–µ–і–µ–ї—М–љ–Њ–≥–Њ –Љ–Њ–љ–Є—В–Њ—А–Є–љ–≥–∞ –Њ–±—Й–µ–≥–Њ –∞–љ–∞–ї–Є–Ј–∞ –Ї—А–Њ–≤–Є –≤¬†—В–µ—З–µ–љ–Є–µ –њ–µ—А–≤—Л—Е —В—А–µ—Е –Љ–µ—Б—П—Ж–µ–≤, –∞¬†–Ј–∞—В–µ–Љ –µ–ґ–µ–Љ–µ—Б—П—З–љ–Њ –≤¬†—Б–≤—П–Ј–Є —Б¬†–≤—Л—Б–Њ–Ї–Є–Љ —А–Є—Б–Ї–Њ–Љ —А–∞–Ј–≤–Є—В–Є—П –∞–≥—А–∞–љ—Г–ї–Њ—Ж–Є—В–Њ–Ј–∞ [58].
–Ъ–≤–µ—В–Є–∞–њ–Є–љ —В–∞–Ї–ґ–µ –Љ–Њ–ґ–µ—В –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞—В—М—Б—П –≤¬†–Ї–∞—З–µ—Б—В–≤–µ –њ—А–µ–њ–∞—А–∞—В–∞ –њ–µ—А–≤–Њ–є –ї–Є–љ–Є–Є, —Е–Њ—В—П –µ–≥–Њ —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В—М, –њ–Њ¬†–Њ—Ж–µ–љ–Ї–∞–Љ, –љ–Є–ґ–µ, —З–µ–Љ —Г¬†–Ї–ї–Њ–Ј–∞–њ–Є–љ–∞ [58]. –Ю–і–љ–∞–Ї–Њ –≤—Б–ї–µ–і—Б—В–≤–Є–µ –ї—Г—З—И–µ–є –њ–µ—А–µ–љ–Њ—Б–Є–Љ–Њ—Б—В–Є –Є¬†–±–Њ–ї–µ–µ –≤—Л—Б–Њ–Ї–Њ–є –±–µ–Ј–Њ–њ–∞—Б–љ–Њ—Б—В–Є –µ–≥–Њ –њ—А–Є–љ–Є–Љ–∞—О—В –і–∞–ґ–µ —З–∞—Й–µ.
–Э–µ–Њ–±—Е–Њ–і–Є–Љ–Њ –Њ—В–Љ–µ—В–Є—В—М, —З—В–Њ –њ—А–Є –С–Я —Б¬†–і–µ–Љ–µ–љ—Ж–Є–µ–є –њ—А–Є–Љ–µ–љ–µ–љ–Є–µ –і–∞–ґ–µ –∞—В–Є–њ–Є—З–љ—Л—Е –љ–µ–є—А–Њ–ї–µ–њ—В–Є–Ї–Њ–≤ —Б–Њ–њ—А—П–ґ–µ–љ–Њ —Б¬†—А–Є—Б–Ї–Њ–Љ —А–∞–Ј–≤–Є—В–Є—П –љ–µ–є—А–Њ–ї–µ–њ—В–Є—З–µ—Б–Ї–Њ–≥–Њ —Б–Є–љ–і—А–Њ–Љ–∞, –њ–Њ—Н—В–Њ–Љ—Г –ї–µ—З–µ–љ–Є–µ –њ—А–µ–њ–∞—А–∞—В–∞–Љ–Є —Н—В–Њ–є¬†–≥—А—Г–њ–њ—Л –і–Њ–ї–ґ–љ–Њ –њ—А–Њ–≤–Њ–і–Є—В—М—Б—П –Њ–±–і—Г–Љ–∞–љ–љ–Њ. –Э–µ–є—А–Њ–ї–µ–њ—В–Є—З–µ—Б–Ї–Є–є —Б–Є–љ–і—А–Њ–Љ —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–С–Я –Є¬†–і–µ–Љ–µ–љ—Ж–Є–µ–є –±—Л–ї –Њ–њ–Є—Б–∞–љ –љ–∞¬†—Д–Њ–љ–µ –ї–µ—З–µ–љ–Є—П —А–Є—Б–њ–µ—А–Є–і–Њ–љ–Њ–Љ, –Њ–ї–∞–љ–Ј–µ–њ–Є–љ–Њ–Љ, –∞—А–Є–њ—А–∞–Ј–Њ–ї–Њ–Љ. –Я—А–µ–њ–∞—А–∞—В—Л —Н—В–Њ–є —Д–∞—А–Љ–∞–Ї–Њ—В–µ—А–∞–њ–µ–≤—В–Є—З–µ—Б–Ї–Њ–є¬†–≥—А—Г–њ–њ—Л –і–Њ–ї–ґ–љ—Л —Б¬†–Њ—Б—В–Њ—А–Њ–ґ–љ–Њ—Б—В—М—О –љ–∞–Ј–љ–∞—З–∞—В—М—Б—П –њ–Њ–ґ–Є–ї—Л–Љ –њ–∞—Ж–Є–µ–љ—В–∞–Љ —Б¬†—Б–µ—А–і–µ—З–љ–Њ-—Б–Њ—Б—Г–і–Є—Б—В–Њ–є –њ–∞—В–Њ–ї–Њ–≥–Є–µ–є, –њ–Њ—Б–Ї–Њ–ї—М–Ї—Г –њ–Њ–≤—Л—И–∞—О—В —А–Є—Б–Ї¬†—Б–Њ—Б—Г–і–Є—Б—В—Л—Е —Б–Њ–±—Л—В–Є–є (–Є—И–µ–Љ–Є—З–µ—Б–Ї–Є–є –Є–љ—Б—Г–ї—М—В, –Є–љ—Д–∞—А–Ї—В –Љ–Є–Њ–Ї–∞—А–і–∞) –Є¬†—Б–Љ–µ—А—В–Є [57].
–Т¬†2016¬†–≥.¬†–≤ –°–®–Р –±—Л–ї –Ј–∞—А–µ–≥–Є—Б—В—А–Є—А–Њ–≤–∞–љ –љ–Њ–≤—Л–є —В–∞–±–ї–µ—В–Є—А–Њ–≤–∞–љ–љ—Л–є –ї–µ–Ї–∞—А—Б—В–≤–µ–љ–љ—Л–є –њ—А–µ–њ–∞—А–∞—В –і–ї—П –ї–µ—З–µ–љ–Є—П¬†–≥–∞–ї–ї—О—Ж–Є–љ–∞—Ж–Є–є –Є¬†–Є–ї–ї—О–Ј–Є–є –њ—А–Є –±–Њ–ї–µ–Ј–љ–Є –Я–∞—А–Ї–Є–љ—Б–Њ–љ–∞¬†вАУ –њ–Є–Љ–∞–≤–∞–љ—Б–µ—А–Є–љ, –Њ–±—А–∞—В–љ—Л–є –∞–≥–Њ–љ–Є—Б—В 5-HT2A-—А–µ—Ж–µ–њ—В–Њ—А–Њ–≤ (–њ–Њ–і—В–Є–њ —Б–µ—А–Њ—В–Њ–љ–Є–љ–Њ–≤—Л—Е —А–µ—Ж–µ–њ—В–Њ—А–Њ–≤). –Я—А–µ–њ–∞—А–∞—В –љ–µ¬†–њ—А–Њ—П–≤–ї—П–µ—В –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –≤¬†–Њ—В–љ–Њ—И–µ–љ–Є–Є –і–Њ—Д–∞–Љ–Є–љ–Њ–≤—Л—Е,¬†–≥–Є—Б—В–∞–Љ–Є–љ–Њ–≤—Л—Е, –Љ—Г—Б–Ї–∞—А–Є–љ–Њ–≤—Л—Е –Є¬†–∞–і—А–µ–љ–Њ—А–µ—Ж–µ–њ—В–Њ—А–Њ–≤. –Ш–Ј—Г—З–µ–љ–Є—О —Н—В–Њ–≥–Њ –њ—А–µ–њ–∞—А–∞—В–∞ –±—Л–ї–Њ –њ–Њ—Б–≤—П—Й–µ–љ–Њ 25¬†–Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є (–Њ–±—Й–µ–µ —З–Є—Б–ї–Њ –Є—Б–њ—Л—В—Г–µ–Љ—Л—Е –Њ–Ї–Њ–ї–Њ 1200). –Т¬†—А–µ–Ј—Г–ї—М—В–∞—В–µ –±—Л–ї–Њ –і–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ –њ–Є–Љ–∞–≤–∞–љ—Б–µ—А–Є–љ —Г–Љ–µ–љ—М—И–∞–µ—В —З–∞—Б—В–Њ—В—Г –Є/–Є–ї–Є —В—П–ґ–µ—Б—В—М¬†–≥–∞–ї–ї—О—Ж–Є–љ–∞—Ж–Є–є –Є¬†–Є–ї–ї—О–Ј–Є–є, –љ–Њ¬†–љ–µ –≤–ї–Є—П–µ—В –љ–∞¬†–≤—Л—А–∞–ґ–µ–љ–љ–Њ—Б—В—М –і–≤–Є–≥–∞—В–µ–ї—М–љ—Л—Е —А–∞—Б—Б—В—А–Њ–є—Б—В–≤ –њ—А–Є –С–Я [56].
–Р—Д—Д–µ–Ї—В–Є–≤–љ—Л–µ —А–∞—Б—Б—В—А–Њ–є—Б—В–≤–∞ –Є¬†–і–µ–њ—А–µ—Б—Б–Є—П¬†вАУ —З–∞—Б—В—Л–µ —Б–Є–Љ–њ—В–Њ–Љ—Л –њ—А–Є –С–Я —Б¬†–і–µ–Љ–µ–љ—Ж–Є–µ–є –Є¬†–±–µ–Ј –љ–µ–µ. –°–Є–Љ–њ—В–Њ–Љ—Л –і–µ–њ—А–µ—Б—Б–Є–Є –љ–µ¬†–≤—Б–µ–≥–і–∞ –ї–µ–≥–Ї–Њ –і–Є–∞–≥–љ–Њ—Б—В–Є—А—Г—О—В—Б—П —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–С–Я¬†вАУ —З–∞—Б—В–Њ –њ–∞—Ж–Є–µ–љ—В –љ–µ¬†–ґ–∞–ї—Г–µ—В—Б—П –љ–∞¬†—Б–љ–Є–ґ–µ–љ–Є–µ –љ–∞—Б—В—А–Њ–µ–љ–Є—П. –Ю–і–љ–Њ–≤—А–µ–Љ–µ–љ–љ–Њ —Б¬†–і–µ–њ—А–µ—Б—Б–Є–µ–є –љ–∞—А–∞—Б—В–∞–µ—В –≤—Л—А–∞–ґ–µ–љ–љ–Њ—Б—В—М –Є¬†–і–≤–Є–≥–∞—В–µ–ї—М–љ—Л—Е, –Є¬†–Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л—Е –љ–∞—А—Г—И–µ–љ–Є–є. –Т¬†—Н—В–Њ–є —Б–≤—П–Ј–Є —Б–Є–Љ–њ—В–Њ–Љ—Л –і–µ–њ—А–µ—Б—Б–Є–Є –љ—Г–ґ–і–∞—О—В—Б—П –≤¬†–њ—А–Є—Ж–µ–ї—М–љ–Њ–є –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–µ –Љ–µ—В–Њ–і–∞–Љ–Є —А–∞—Б—Б–њ—А–Њ—Б–∞ –Є¬†–∞–љ–Ї–µ—В–Є—А–Њ–≤–∞–љ–Є—П –љ–µ¬†—В–Њ–ї—М–Ї–Њ –њ–∞—Ж–Є–µ–љ—В–Њ–≤, –љ–Њ¬†–Є –Є—Е —А–Њ–і—Б—В–≤–µ–љ–љ–Є–Ї–Њ–≤.
–Ъ–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –Є¬†–Љ–µ—В–∞–∞–љ–∞–ї–Є–Ј —А–∞–Ј–ї–Є—З–љ—Л—Е –њ—А–µ–њ–∞—А–∞—В–Њ–≤, –њ—А–Є–Љ–µ–љ—П–≤—И–Є—Е—Б—П –≤¬†–ї–µ—З–µ–љ–Є–Є –і–µ–њ—А–µ—Б—Б–Є–Є —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–С–Я –Є¬†–і–µ–Љ–µ–љ—Ж–Є–µ–є, —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г—О—В –Њ –љ–∞–Є–±–Њ–ї—М—И–µ–є –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –і–µ–Ј–Є–њ—А–∞–Љ–Є–љ–∞ –Є¬†—Ж–Є—В–∞–ї–Њ–њ—А–∞–Љ–∞, –∞¬†—В–∞–Ї–ґ–µ –њ–∞—А–Њ–Ї—Б–µ—В–Є–љ–∞ –Є¬†–≤–µ–љ–ї–∞—Д–∞–Ї—Б–Є–љ–∞ –њ–Њ¬†—Б—А–∞–≤–љ–µ–љ–Є—О —Б¬†–і—А—Г–≥–Є–Љ–Є –∞–љ—В–Є–і–µ–њ—А–µ—Б—Б–∞–љ—В–∞–Љ–Є –Є¬†–њ–ї–∞—Ж–µ–±–Њ [59]. –Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, –і–ї—П –ї–µ—З–µ–љ–Є—П –і–µ–њ—А–µ—Б—Б–Є–Є —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–С–Я –Є¬†–і–µ–Љ–µ–љ—Ж–Є–µ–є –њ—А–µ–і–њ–Њ—З—В–Є—В–µ–ї—М–љ–µ–µ –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞—В—М —Б–µ–ї–µ–Ї—В–Є–≤–љ—Л–µ –Є–љ–≥–Є–±–Є—В–Њ—А—Л –Њ–±—А–∞—В–љ–Њ–≥–Њ –Ј–∞—Е–≤–∞—В–∞ —Б–µ—А–Њ—В–Њ–љ–Є–љ–∞ –Є¬†—Б–µ–ї–µ–Ї—В–Є–≤–љ—Л–µ –Є–љ–≥–Є–±–Є—В–Њ—А—Л –Њ–±—А–∞—В–љ–Њ–≥–Њ –Ј–∞—Е–≤–∞—В–∞ —Б–µ—А–Њ—В–Њ–љ–Є–љ–∞ –Є¬†–љ–Њ—А–∞–і—А–µ–љ–∞–ї–Є–љ–∞.
N.V. Trofimova, I.S. Preobrazhenskaya, M.A. Bykanova
I.M. Sechenov First Moscow State Medical University (Sechenovskiy University)
Contact person: Irina Sergeyevna Preobrazhenskaya, irinasp2@yandex.ru
The article provides the current data on the epidemiology, morphology, clinical manifestations of Parkinson's disease combined with dementia. On discussion different points of view about the nature of dementia in Parkinson's disease as well as the possible contribution to the development of vascular dementia and neurodegenerative diseases. The modern diagnostic criteria for dementia in Parkinson's disease are provided. The recommendation data and clinical studies of the efficacy of drugs used in the treatment of motor, cognitive, psychiatric disorders in patients with combination of dementia and Parkinson's disease are summarized.
–£–≤–∞–ґ–∞–µ–Љ—Л–є –њ–Њ—Б–µ—В–Є—В–µ–ї—М uMEDp!
–£–≤–µ–і–Њ–Љ–ї—П–µ–Љ –Т–∞—Б –Њ —В–Њ–Љ, —З—В–Њ –Ј–і–µ—Б—М —Б–Њ–і–µ—А–ґ–Є—В—Б—П –Є–љ—Д–Њ—А–Љ–∞—Ж–Є—П, –њ—А–µ–і–љ–∞–Ј–љ–∞—З–µ–љ–љ–∞—П –Є—Б–Ї–ї—О—З–Є—В–µ–ї—М–љ–Њ –і–ї—П —Б–њ–µ—Ж–Є–∞–ї–Є—Б—В–Њ–≤ –Ј–і—А–∞–≤–Њ–Њ—Е—А–∞–љ–µ–љ–Є—П.
–Х—Б–ї–Є –Т—Л –љ–µ —П–≤–ї—П–µ—В–µ—Б—М —Б–њ–µ—Ж–Є–∞–ї–Є—Б—В–Њ–Љ –Ј–і—А–∞–≤–Њ–Њ—Е—А–∞–љ–µ–љ–Є—П, –∞–і–Љ–Є–љ–Є—Б—В—А–∞—Ж–Є—П –љ–µ –љ–µ—Б–µ—В –Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ—Б—В–Є –Ј–∞ –≤–Њ–Ј–Љ–Њ–ґ–љ—Л–µ –Њ—В—А–Є—Ж–∞—В–µ–ї—М–љ—Л–µ –њ–Њ—Б–ї–µ–і—Б—В–≤–Є—П, –≤–Њ–Ј–љ–Є–Ї—И–Є–µ –≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ —Б–∞–Љ–Њ—Б—В–Њ—П—В–µ–ї—М–љ–Њ–≥–Њ –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є—П –Т–∞–Љ–Є –Є–љ—Д–Њ—А–Љ–∞—Ж–Є–Є —Б –њ–Њ—А—В–∞–ї–∞ –±–µ–Ј –њ—А–µ–і–≤–∞—А–Є—В–µ–ї—М–љ–Њ–є –Ї–Њ–љ—Б—Г–ї—М—В–∞—Ж–Є–Є —Б –≤—А–∞—З–Њ–Љ.
–Э–∞–ґ–Є–Љ–∞—П –љ–∞ –Ї–љ–Њ–њ–Ї—Г ¬Ђ–Т–Њ–є—В–Є¬ї, –Т—Л –њ–Њ–і—В–≤–µ—А–ґ–і–∞–µ—В–µ, —З—В–Њ —П–≤–ї—П–µ—В–µ—Б—М –≤—А–∞—З–Њ–Љ –Є–ї–Є —Б—В—Г–і–µ–љ—В–Њ–Љ –Љ–µ–і–Є—Ж–Є–љ—Б–Ї–Њ–≥–Њ –≤—Г–Ј–∞.