Патогенетическое обоснование антиоксидантной терапии при хронической ишемии мозга
- Аннотация
- Статья
- Ссылки


В соответствии с Классификацией ВОЗ цереброваскулярных болезней (1981) и отечественной Классификацией сосудистого поражения головного и спинного мозга Института неврологии РАМН нарушения мозгового кровообращения могут быть хроническими (медленно прогрессирующими) и острыми (2).
Хроническая недостаточность мозгового кровообращения (ХНМК), или хроническая сосудисто-мозговая недостаточность (ХСМН) – наиболее распространенная цереброваскулярная патология, возникающая обычно на фоне общих сердечно-сосудистых заболеваний. Чаще это атеросклероз, гипертоническая болезнь и их сочетания, сахарный диабет, возможны васкулиты при системных заболеваниях соединительной ткани и другие заболевания, сопровождающиеся поражением сосудов, болезни крови, ведущие к увеличению ее вязкости (эритремия, макроглобулинемия, криоглобулинемия и др.).
Отмеченные выше, а также некоторые другие формы патологии могут привести к изменениям общей и локальной гемодинамики с проявлениями периодических, а в дальнейшем и постепенно прогрессирующих хронических сосудисто-мозговых патологий.
В настоящее время выделяются следующие стадии ХСМН: начальные проявления недостаточности мозгового кровообращения и дисциркуляторная энцефалопатия (3). При этом начальные проявления недостаточности кровоснабжения мозга обычно возникают, если приток крови к мозгу меньше 45-30 мл/100 г/мин.; дисциркуляторная энцефалопатия развивается при постоянном поступлении крови в мозг в пределах 35-20 мл/100 г/мин. Большинством авторов критическим признается регионарный кровоток в пределах 19 мл/100 г/мин. (функциональный порог кровоснабжения мозга), при котором уже оказываются нарушенными функции соответствующих участков мозга. Процесс гибели нервных клеток происходит при регионарном артериальном мозговом кровотоке, сниженном до 8-10 мл/10 мг/мин. (инфарктный порог кровоснабжения мозга). Прекращение в головном мозге кровотока на 5-8 мин. ведет к развитию в нем необратимых изменений (2, 3).
Термин «дисциркуляторная энцефалопатия» был предложен Г.А. Максудовым и В.М. Коганом в 1958 г. и позднее включен в отечественную классификацию поражений головного и спинного мозга. В МКБ-10 (1995), как и в прежней Международной классификации болезней девятого пересмотра, этот термин отсутствует. Среди возможных близких по клинической картине состояний в МКБ-10 упоминается церебральный атеросклероз, прогрессирующая сосудистая лейкоэнцефалопатия, гипертензивная энцефалопатия, другие уточненные поражения сосудов мозга, в том числе ишемия мозга (хроническая), цереброваскулярная болезнь неуточненная. Наибольшее развитие этот вопрос получил в трудах отечественных неврологов. Некоторыми из них предлагались иные обозначения этого состояния – ишемическая болезнь (головного) мозга (3), сосудистая энцефалопатия, или ангиоэнцефалопатия (2). При описании поздних, далеко зашедших стадий сосудистой патологии головного мозга за рубежом используется термин «сосудистая деменция» (20), который отражает одно из ярких клинических проявлений этого страдания, или с учетом патоморфологической основы этой патологии, «лакунарное состояние» (15).
Изучение структурных основ ишемического повреждения мозга при сочетании различных этиопатогенетических факторов позволило перейти от синдромологического термина «дисциркуляторная энцефалопатия» к понятию хроническая ишемия мозга (ХИМ) – недифференцированной форме хронической сосудистой патологии мозга. ХИМ – это состояние, проявляющееся прогрессирующим многоочаговым расстройством функций головного мозга, обусловленным недостаточностью церебрального кровообращения (9).
Патогенез ХИМ обусловлен недостаточностью мозгового кровообращения в относительно стабильной ее форме и (или) повторными эпизодами дисциркуляции, которые протекают с явной клинической симптоматикой (в виде инсульта или транзиторной ишемической атаки) или субклинически (3, 5, 9). Любопытно отметить, что почти у 80% пожилых лиц с выявленными при аутопсии инфарктами при жизни указаний на перенесенный инсульт не было (4). Патологические изменения сосудистой стенки вследствие артериальной гипертонии, атеросклероза и др. вызывают нарушение ауторегуляции мозгового кровообращения. В свою очередь возникает все большая его зависимость от состояния системной гемодинамики, также оказывающейся нестабильной вследствие тех же заболеваний сердечно-сосудистой системы. К этому добавляется нарушение нейрогенной регуляции системной и церебральной гемодинамики. Сама же по себе гипоксия мозга приводит к дальнейшему повреждению механизмов ауторегуляции мозгового кровообращения.
Изучение состояния сосудистой системы мозга вне- и внутричерепных артерий показало, что при разном характере основного заболевания изменения, сопровождающие ишемию мозга, неодинаковы. Обусловленная атеросклерозом патология сосудов головного мозга является наиболее частой причиной развития ишемических нарушений мозгового кровообращения, которые занимают основное место в структуре сосудистой патологии мозга (2).
На определенном этапе эволюции и прогрессирования атеросклеротической ангиопатии головного мозга возникает существенное снижение или прекращение локального мозгового кровотока. Это приводит к очаговой или распространенной гипоксии и ишемии мозга со структурными изменениями его, различными по локализации, тяжести и протяженности. Возникает картина атеросклеротической ангиоэнцефалопатии, которая клинически проявляется в виде определенных симптомов и синдромов.
Таким образом, церебральный атеросклероз, до определенной стадии своего развития протекавший бессимптомно, становится клинически значимым.
Пусковым механизмом повреждающего действия ишемии на мозг является снижение уровня высокоэнергетических фосфатов. Недостаток кислорода стимулирует переход на анаэробный гликолиз, обеспечивающий сохранение возможности синтеза АТФ и приводящий к накоплению молочной кислоты, что ведет к выраженному лактоацидозу. Это проявляется в снижении величины рН в ткани мозга. Величина рН влияет на регуляцию локального мозгового кровотока, который в свою очередь обеспечивает доставку О2 к ткани. Величина рН определяет функционирование клеточных мембран и активность ферментов, участвующих в гликолизе. Установлено, что в условиях неполной ишемии или при попытке неадекватной реперфузии происходит дальнейшее снабжение мозга энергетическим субстратом (глюкозой) для анаэробного гликолиза, а это ведет к усилению лактоацидоза и углублению поражения нейронов (21).
Истощение энергетического субстрата приводит к нарушению функции Ка+/Na+-насоса, возникает деполяризация клеточных мембран, нарушается их проницаемость. Ионы кальция в значительных количествах поступают из внеклеточной жидкости внутрь клеток. Включаются дополнительные повреждающие механизмы, в том числе высвобождение свободных жирных кислот. Происходит накопление свободных радикалов, стимулирующих процессы перекисного окисления липидов, что приводит к быстрой гибели нейронов. В процессе развития ишемии избирательно нарушаются механизмы синаптической передачи, увеличивается внеклеточная концентрация γ-аминомасляной кислоты и глутамата. В ишемизированной ткани уменьшается синтез дофамина и норадреналина, а высвобождение серотонина намного возрастает. Все это приводит к нарушению ауторегуляции мозгового кровотока, развитию вазоспазма, усилению агрегации тромбоцитов и формированию внутрисосудистого стаза, что в свою очередь углубляет ишемию, делает ее необратимой. Кроме того, высвобождение катехоламинов на пресинаптическом уровне может, по-видимому, вызвать усиление активности нейронов и возникновение дополнительных потребностей в энергетическом субстрате, что в условиях его дефицита при ишемии углубляет ишемическое поражение. Также в процессе перекисного окисления липидов повышается образование простаноидов и лейкотриенов. В конечном итоге это ведет к изменениям сосудистой реактивности, нарушению проницаемости сосудов, повышении агрегационной способности тромбоцитов, т.е. к еще большему углублению ишемии (16).
В процессе необратимого изменения структур клетки при ишемии накопление свободных радикалов, продуктов перекисного окисления липидов и эйкозаноидов является одним из ключевых моментов. Вместе с тем, для мозга характерно низкое содержание основных компонентов антиоксидантной защиты. В целом, именно дефицит антиоксидантной системы в мозговой ткани объясняет ее особую чувствительность к продукции свободнорадикальных соединений (1, 8, 19). Так, составляя всего 2% от общей массы тела, мозг утилизирует 20-25% получаемого кислорода, поэтому переход в свободно-радикальную форму даже 0,1% метаболизируемого нейронами кислорода окажется токсичным для мозговой ткани.
Механизм перекисного окисления липидов (ПОЛ) в клетках ЦНС аналогичен механизмам в других тканях, однако интенсивность процесса здесь значительно выше (14). Во многом это определяется высоким содержанием в мозге полиненасыщенных жирных кислот – субстратов ПОЛ. Так, содержание фосфолипидов (ФЛ) в мозге в 1,5 раза больше, чем в печени и в 3-4 раза больше, чем в сердце. Высокая интенсивность ПОЛ в ЦНС определяется также высокими концентрациями ионов металлов с переменной валентностью, необходимых для функционирования ферментов и работы дофаминовых рецепторов (1, 19). Нарушения кислородного метаболизма, приводящего к ацидозу, способствует высвобождению ионов металлов, становящихся катализаторами свободнорадикальных реакций (14). Таким образом, продукция СР в нейронах может усиливаться в результате различных неблагоприятных факторов, а также при изменении условий функционирования или генетически детерминированных дефектах клетки. Наряду с увеличением концентрации субстратов ПОЛ в зоне ишемии нарастает интенсивность генерации активных форм кислорода (АФК), накапливаются прооксиданты – стимуляторы ПОЛ. Эти условия сочетаются со снижением активности антиоксидантных ферментов и нарушением функции физиологических систем защиты. Очаговая ишемия характеризуется продукцией СР, высвобождением глутамата из очага заболевания, который диффузно переходит в зону ишемической полутени, увеличением в поврежденных нейронах ионов Са2+, активацией синтеза NO. В дальнейшем происходит индукция NO-синтазы в эндотелии и лейкоцитах, активация ЦОГ-2 в нейронах и повреждение митохондрий. Ишемия и последующее возобновление кровотока (реоксигенация) – процессы, которые приводят к генерации АФК. В случае невозможности нейтрализовать избыток АФК, генерируемых в условиях ишемии мозга, происходит активация каскада реакций, вызывающих хаотическую (по типу некроза) или программируемую (по типу апоптоза) смерть клетки (5, 13).
Вместе с тем, активация ПОЛ наблюдается не только при остром ишемическом повреждении мозга и реперфузии, когда этот процесс развивается лавинообразно и приводит к развитию окислительного стресса. Повышение концентрации продуктов ПОЛ возможно также у больных с хронической недостаточностью мозгового кровообращения. Состояние анти- и прооксидантной системы в процессе старения является одним из патогенетических факторов, определяющих вероятность развития различных патологических состояний, в частности сосудистой деменции, развитие которой обусловлено снижением мозгового кровотока и, соответственно, развитием гипоксии тканей, которые приводят к развитию окислительного стресса (6, 7).
В нарушении СРП у больных хронической ишемией мозга можно выделить начальную стадию, когда нарушение кровотока и гипоксия приводят к интенсивной генерации АФК и мобилизации АО системы. С нарастанием гипоксии мозга, интенсификацией генерации АФК происходит истощение эндогенных антиоксидантных ресурсов, в результате чего выраженные изменения качественного и количественного липидного и белкового составов клеточных мембран, окислительная деструкция ДНК, дисбаланс АОА системы могут привести к интенсивному апоптозу нейронов (5, 8).
Таким образом, выраженная активация свободнорадикальных процессов является одним из ключевых патогенетических механизмов развития и прогрессирования хронических цереброваскулярных заболеваний. Поэтому при разработке подходов к терапии хронической ишемии мозга встает вопрос об адекватной коррекции нарушений процессов свободнорадикального окисления, направленной на усиление антиоксидантной защитной системы организма (10).
Перспективным антиоксидантом в профилактике и лечении ишемических, возрастных и нейродегенеративных заболеваний мозга является α-липоевая кислота (α-ЛК) – тиоловое соединение с прямым антиоксидантным действием (синонимы – тиоктовая, липоновая кислота, витамин N). Впервые α-липоевая кислота выделена в кристаллическом виде из говяжьей печени в 1951 году, а в 1953 создан ее синтетический аналог (12). Тиоловые соединения способны накапливаться в мозге и обладают выраженным антиоксидантным защитным действием в условиях гипоксии и ишемии. α-Липоевая кислота является коферментом, входящим в состав энзимов группы кокарбоксилаз. В организме α-ЛК образует динамичную окислительно-восстановительную систему, которая участвует в переносе ацильных групп в составе многокомпонентных ферментных систем. Основное значение имеет ее участие в качестве кофактора в окислительном декарбоксилировании α-кетокислот (пировиноградной и кетоглутаровой), протекающем в матриксе митохондрий. α-ЛК играет значительную роль в процессе образования энергии в организме. Она облегчает превращение молочной кислоты в пировиноградную с последующим ее декарбоксилированием, т.е. способствует ликвидации метаболического ацидоза. Способствуя образованию коэнзима А (КоА), она облегчает перенос ацетата и жирных кислот из цитозоля в матрикс митохондрий для последующего окисления. Это сопровождается уменьшением выраженности жировой дистрофии гепатоцитов, активизацией метаболической функции печени и желчеотделения, нередко этому сопутствует и снижение содержания липидов в плазме крови. Кроме того, α-ЛК оказывает липотропное действие, ускоряет окисление жирных кислот. R-изомер ЛК увеличивает захват глюкозы на периферии. Совместно с инсулином способствует перемещению в мембрану клетки глюкозтранспортирующих протеинов, что обеспечивает 20-40-кратное поступление глюкозы в клетки инсулинозависимых тканей (18). Однако «второе рождение» α-липоевой кислоты для медицинской практики в большей мере связано с ее антиоксидантным действием, который обусловлен наличием двух тиоловых групп в ее молекуле (отсюда приставка «тио»), а также способностью связывать молекулы радикалов и свободное тканевое железо (предотвращая его участие в ПОЛ).
Антиоксидантная активность α-липоевой кислоты используется во многих областях медицины. Получены доказательства того, что она не только обладает самостоятельным антиоксидантным потенциалом, но и обеспечивает мощную поддержку работы других антиоксидантных звеньев в организме (6, 11). В этом отношении ее протективное действие тесно связано с гомеостазом в системе глутатиона и убихинона. Принимая на себя 2 электрона убихинона и превращаясь в дигидролипоевую кислоту, α-липоевая кислота поддерживает коэнзим Q в «рабочем состоянии». Эффективно использование тиоктовой кислоты в комбинации с коэнзимом Q. Рекомендуется назначение α-липоевой кислоты (совместно с другими антиоксидантами) в терапии врожденных форм гемолитических анемий (серповидно-клеточной, талассемии, анемии на фоне врожденной недостаточности глюкозо-6-фосфатдегидрогеназы), поскольку при этих состояниях повышена активность прооксидантного звена. α-ЛК оказывает прямое стимулирующее действие на активность уропорфириноген-декарбоксилазы, что может быть использовано в лечении порфирий. Важное место в клинической практике в настоящее время занимает использование α-липоевой кислоты в лечении больных сахарным диабетом. Активно участвуя в метаболических процессах, α-ЛК способна оказывать детоксицирующее действие. В частности, α-ЛК выполняет роль антидота при отравлении солями ртути, акриламидом, при отравлении свинцом (12). Тиоктовая кислота может способствовать устранению побочных эффектов ряда лекарственных препаратов. В частности, назначение цисплатина, гентамицина, амикацина сопровождается активизацией ПОЛ, снижением запасов глутатиона в клетках улитки внутреннего уха, нарастанием уровня МДА. Показано, что назначение антиоксидантов, и в частности α-липоевой кислоты (100 мг/кг в день) уменьшает выраженность ототоксического воздействия (17). Кроме того что тиоктовая кислота подавляет синтез NO гепатоцитами при септическом шоке, в лечении поражения печени вирусом гепатита С ингибирует острую фазу воспаления и болевой синдром в этой фазе (11, 18).
Широкий терапевтический потенциал α-липоевой кислоты, влияющий на энергетический метаболизм и редукцию окислительного стресса, является патогенетическим обоснованием для использования этого средства у больных с ишемией мозга.
На кафедре неврологии Российского государственного медицинского университета совместно с НИИ физико-химической медицины ФМБА РФ проведены исследования препарата Берлитион®300 (Берлин-Хеми АГ/Менарини Групп, Германия) созданного на основе α-липоевой кислоты, в качестве нейропротективной терапии хронической ишемии мозга (ХИМ) для коррекции свободнорадикальных процессов. Целью нашей работы явилось проведение комплексного клинико-биохимического исследования эффективности препарата Берлитион®300 у больных с хроническими цереброваскулярными заболеваниями, с анализом взаимосвязи между клинической эффективностью препарата и количественными характеристиками окислительного стресса. В задачи исследования входила оценка динамики основных клинических синдромов и свободнорадикальных процессов на фоне лечения препаратом Берлитион®300.
В исследование были включены 53 пациента с хронической ишемией мозга во II и III стадиях заболевания, в том числе с последствиями ишемического инсульта. Диагноз ХИМ устанавливался согласно Международной классификации болезней десятого пересмотра на основании тяжести клинического синдрома при разных стадиях ХИМ и подтверждался данными МРТ-исследования.
Возраст исследуемых составлял от 49 до 78 лет (средний возраст составил 61,2 ± 8,8 лет). Из 53 пациентов 33 – женщины, 20 – мужчины. Эффективность препарата Берлитион® 300 исследовалась открытым способом. Методом случайной выборки больные были рандомизированы в 2 группы. Пациенты I группы (33 человека) получали препарат Берлитион®300 в суточной дозе 300 ЕД раствора, разведенного в 200 мл 0,9% физиологического раствора, курс составил 14 дней. Базовая терапия включала гипотензивные, антиагрегантные, кардиальные и противодиабетические препараты при условии, что их прием начинался до начала обследования, а дозировки на протяжении периода лечения оставались неизменными. Группу сравнения составили 20 пациентов, сопоставимых по полу, возрасту и характеру заболевания, получавших в течение 14 дней вышеуказанную базовую терапию.
Исследование неврологического статуса с оценкой общемозговых, астеноневротических, двигательных, вестибуло-мозжечковых, экстрапирамидных, чувствительных и псевдобульбарных расстройств проводилось по адаптированной количественной неврологической шкале А.И. Федина со стандартизированными критериями оценки выраженности каждого симптома.
Оценка перекисного окисления липидов до и после лечения проводилась по данным спектрофотометрического измерения уровня малонового диальдегида (МДА), полученного с помощью реакции с тиобарбитуровой кислотой (ТБК-реактивные продукты) через 24 часа после медь-индуцированного окисления плазмы. Измеряемый параметр характеризует уровень окисляемости плазмы, а его повышение у больных хронической ишемией свидетельствует о нестабильном состоянии и может расцениваться как маркер прогрессирования ишемического процесса. Следует отметить, что, так как при активации СРП окислительной модификации в плазме подвергаются и липиды, и белки, окислительную резистентность плазмы оценивали не только по накоплению продуктов окисления липидов, но и белков. В работе были использованы следующие подходы: определение общего количества окисленных белков по накоплению карбонильных продуктов окисления белков (ДНФГ), определение функционального состояния и окисляемости сывороточного альбумина с помощью флуоресцентного зонда К-35, определение с помощью флуоресцентной метки на SH-группы ТиоГло ТМ5 соединений с SH-группами, которые обладают антиоксидантными свойствами.
Результаты исследований показали, что у пациентов ХИМ в фоновом состоянии регистрировался повышенный уровень МДА через 24 часа после медь-индуцированного окисления плазмы (МДАо), высокий уровень содержания карбонильных продуктов окисления белков, снижение содержания SH-групп и связывающей способности альбумина. Данные свидетельствовали об активизации процессов свободно-радикального повреждения и снижении ресурсов антиоксидантной защиты у данного контингента больных.
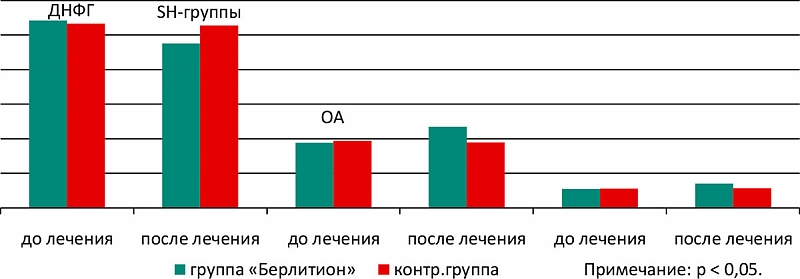
По завершении курса лечения выявлено статистически значимое уменьшение выраженности как субъективных, так и объективных симптомов заболевания во всех исследуемых группах. Однако, разница суммы баллов, отражающая тяжесть субъективных проявлений заболевания до и после лечения в I группе больных, получавших Берлитион®300, оказалась достоверно большей по сравнению с контрольной группой. При общей оценке эффекта лечения было выявлено четкое превалирование положительных результатов при использовании препарата Берлитион®300 (93,2%) по сравнению с группой сравнения (60%) (рисунок 1). В то же время при межгрупповом сравнении динамики органических симптомов заболевания значимых различий выявлено не было. Клинический эффект лечения проявлялся снижением выраженности эмоционально-волевых расстройств, улучшением функции памяти и внимания, уменьшением выраженности цефалгического, вестибуло-мозжечкового, кохлео-вестибулярного и астенических синдромов. Клинический эффект препарата коррелировал с параметрами ПОЛ. Так, у больных I группы выявлено достоверное повышение окислительной устойчивости плазмы, т.е. снижение уровней МДАо, карбонильных соединений, увеличение количества восстановленных SH-групп, повышение связывающей способности альбумина – его окисляемости (ОА), в то время как в группе контроля динамики практически не наблюдалось (рисунок 1, 2).
Анализ результатов исследования выявил статистически значимую зависимость между степенью регресса неврологических симптомов в баллах и исходным уровнем активности СРП в группе больных, получавших Берлитион®300 (r = 0,44, p = 0,02), в то время как в группе сравнения подобной зависимости не было выявлено.
Отмечена также тенденция к обратной корреляции между степенью клинического улучшения в результате лечения препаратом Берлитион®300 и уровнем повышения активности СРП (r = -0,37, p = 0,053). Вместе с тем, несмотря на проведенную терапию, исследуемые параметры перекисного окисления липопротеинов хотя и имели достоверный уровень положительных изменений, но к 15-м суткам не достигали значений возрастных границ нормы, что послужило основанием продолжить курс лечения препаратом Берлитион®300 для достижения еще более выраженного корригирующего эффекта. Из группы больных (30 человек), получавших Берлитион®300 в инъекционной форме, 20 пациентам (подгруппа 1) было продолжено лечение препаратом в таблетированной форме по 300 ЕД 2 раза в сутки в течение 4 недель, 10 пациентов этой группы были включены в подгруппу 2 (сравнения).
Результаты исследования (рисунок 3) показали, что через 15 дней продолженного лечения в подгруппе 1 выявлялась дальнейшая тенденция к снижению уровней МДАо, карбонильных соединений, увеличению количества восстановленных SH-групп, повышению связывающей способности альбумина, а к 30-му дню эти показатели приблизились к значениям группы из условно здоровых лиц (донорам). В подгруппе 2 дальнейшей коррекции показателей активности СРП и АО системы не выявлялось. Кроме того, межгрупповое сравнение динамики органических симптомов заболевания выявило значимые различия в подгруппах 1 и 2, что свидетельствовало о большем клиническом эффекте у больных, получавших лечение препаратом Берлитион®300 в течение 6 недель.
Анализ полученных данных позволил сделать вывод о том, что у больных хронической ишемией мозга регистрируется активизация процессов перекисного окисления липопротеинов, требующая медикаментозной коррекции. Нейропротективное действие препарата Берлитион®300, связанное с предотвращением повреждающего воздействия свободных радикалов на клеточные мембраны и уменьшением выраженности окислительного стресса, является патогенетическим обоснованием использования его как антиоксидантного средства при ишемическом поражении головного мозга. Критерием эффективности лечения может служить оценка окислительной устойчивости плазмы по накоплению продуктов окисления липидов (МДА), белков (карбонилов) и соединений с SH-группами, которые обладают антиоксидантными свойствами.
Эффективность действия антиоксиданта, как и других лекарственных веществ, определяется дозой, сроками и способами их введения. В связи с этим мы рекомендуем для коррекции свободнорадикальных процессов при хронической ишемии мозга курсовое использование инъекционной и таблетированной форм α-липоевой кислоты: в начальной инъекционной дозе 300 ЕД/сут. в течение 2 недель с последующим переходом на таблетированную форму 600 ЕД/сут. в течение 4 недель для стабилизации клинического и антиоксидантного эффектов. Отчетливое нейропротективное действие препарата Берлитион®300 позволяет рекомендовать его для курсового лечения хронической цереброваскулярной патологии.
Уважаемый посетитель uMEDp!
Уведомляем Вас о том, что здесь содержится информация, предназначенная исключительно для специалистов здравоохранения.
Если Вы не являетесь специалистом здравоохранения, администрация не несет ответственности за возможные отрицательные последствия, возникшие в результате самостоятельного использования Вами информации с портала без предварительной консультации с врачом.
Нажимая на кнопку «Войти», Вы подтверждаете, что являетесь врачом или студентом медицинского вуза.