–°–ĺ–≤—Ä–Ķ–ľ–Ķ–Ĺ–Ĺ–ĺ–Ķ —Ā–ĺ—Ā—ā–ĺ—Ź–Ĺ–ł–Ķ –Ņ—Ä–ĺ–Ī–Ľ–Ķ–ľ—č –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź –ī–ł—Ā—Ā–Ķ–ľ–ł–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–≥–ĺ —Ä–į–ļ–į –Ņ–ĺ–ī–∂–Ķ–Ľ—É–ī–ĺ—á–Ĺ–ĺ–Ļ –∂–Ķ–Ľ–Ķ–∑—č –ł –≤–ĺ–∑–ľ–ĺ–∂–Ĺ—č–Ķ –Ņ–Ķ—Ä—Ā–Ņ–Ķ–ļ—ā–ł–≤—č
- –ź–Ĺ–Ĺ–ĺ—ā–į—Ü–ł—Ź
- –°—ā–į—ā—Ć—Ź
- –°—Ā—č–Ľ–ļ–ł
–í–≤–Ķ–ī–Ķ–Ĺ–ł–Ķ
–†–į–ļ –Ņ–ĺ–ī–∂–Ķ–Ľ—É–ī–ĺ—á–Ĺ–ĺ–Ļ –∂–Ķ–Ľ–Ķ–∑—č (–†–ü–Ė)¬†‚Äď –Ĺ–Ķ—Ä–Ķ—ą–Ķ–Ĺ–Ĺ–į—Ź –Ņ—Ä–ĺ–Ī–Ľ–Ķ–ľ–į —Ā–ĺ–≤—Ä–Ķ–ľ–Ķ–Ĺ–Ĺ–ĺ–Ļ –ĺ–Ĺ–ļ–ĺ–Ľ–ĺ–≥–ł–ł, —ā–į–ļ –ļ–į–ļ –Ĺ–ł –ĺ–ī–ł–Ĺ –ł–∑ –ľ–Ķ—ā–ĺ–ī–ĺ–≤ –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź –Ĺ–Ķ –Ņ–ĺ–∑–≤–ĺ–Ľ—Ź–Ķ—ā –ī–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ķ –≤—Ä–Ķ–ľ—Ź –ļ–ĺ–Ĺ—ā—Ä–ĺ–Ľ–ł—Ä–ĺ–≤–į—ā—Ć –Ī–ĺ–Ľ–Ķ–∑–Ĺ—Ć. –ė–∑ 230 000 –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö, –∑–į—Ä–Ķ–≥–ł—Ā—ā—Ä–ł—Ä–ĺ–≤–į–Ĺ–Ĺ—č—Ö –≤¬†–ľ–ł—Ä–Ķ, —É–ľ–ł—Ä–į—é—ā 98%. –ó–į –Ņ–ĺ—Ā–Ľ–Ķ–ī–Ĺ–ł–Ķ 5 –Ľ–Ķ—ā –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ķ–ľ–ĺ—Ā—ā—Ć –†–ü–Ė –≤–ĺ–∑—Ä–ĺ—Ā–Ľ–į –Ĺ–į 4,2% —Ā—Ä–Ķ–ī–ł –ľ—É–∂—á–ł–Ĺ –ł¬†–Ĺ–į 12,1% —Ā—Ä–Ķ–ī–ł –∂–Ķ–Ĺ—Č–ł–Ĺ. –í¬†–†–ĺ—Ā—Ā–ł–ł –≤¬†2007 –≥. –ļ–ĺ–Ľ–ł—á–Ķ—Ā—ā–≤–ĺ –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö —Ā¬†–≤–Ņ–Ķ—Ä–≤—č–Ķ —É—Ā—ā–į–Ĺ–ĺ–≤–Ľ–Ķ–Ĺ–Ĺ—č–ľ –ī–ł–į–≥–Ĺ–ĺ–∑–ĺ–ľ —Ā–ĺ—Ā—ā–į–≤–ł–Ľ–ĺ 14 037 (–≤ —Ā—ā—Ä—É–ļ—ā—É—Ä–Ķ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ķ–ľ–ĺ—Ā—ā–ł –†–ü–Ė –∑–į–Ĺ—Ź–Ľ 4-–Ķ –ľ–Ķ—Ā—ā–ĺ —Ā—Ä–Ķ–ī–ł –ĺ–Ņ—É—Ö–ĺ–Ľ–Ķ–Ļ –Ņ–ł—Č–Ķ–≤–į—Ä–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ļ —Ā–ł—Ā—ā–Ķ–ľ—č), –į¬†—Ā–ľ–Ķ—Ä—ā–Ĺ–ĺ—Ā—ā—Ć –ĺ—ā –†–ü–Ė¬†‚Äď 14 473 —Ā–Ľ—É—á–į—Ź (–Ņ—Ä–Ķ–≤–į–Ľ–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ —Ā–ľ–Ķ—Ä—ā–Ĺ–ĺ—Ā—ā–ł –Ĺ–į–ī –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ķ–ľ–ĺ—Ā—ā—Ć—é –ĺ–Ī—É—Ā–Ľ–ĺ–≤–Ľ–Ķ–Ĺ–ĺ –Ņ–ĺ—Ā–ľ–Ķ—Ä—ā–Ĺ–ĺ–Ļ –ī–ł–į–≥–Ĺ–ĺ—Ā—ā–ł–ļ–ĺ–Ļ –Ī–ĺ–Ľ–Ķ–∑–Ĺ–ł) [1, 2]. –ē–≤—Ä–ĺ–Ņ–Ķ–Ļ—Ā–ļ–ł–Ķ —Ā—ā–į—ā–ł—Ā—ā–ł—á–Ķ—Ā–ļ–ł–Ķ –ī–į–Ĺ–Ĺ—č–Ķ –Ņ–ĺ –†–ü–Ė –į–Ĺ–į–Ľ–ĺ–≥–ł—á–Ĺ—č —Ä–ĺ—Ā—Ā–ł–Ļ—Ā–ļ–ł–ľ: –≤¬†2008 –≥. –Ņ–ĺ–ļ–į–∑–į—ā–Ķ–Ľ—Ć –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ķ–ľ–ĺ—Ā—ā–ł (68 500 —Ā–Ľ—É—á–į–Ķ–≤) –Ī—č–Ľ –ľ–Ķ–Ĺ—Ć—ą–Ķ –Ņ–ĺ–ļ–į–∑–į—ā–Ķ–Ľ—Ź —Ā–ľ–Ķ—Ä—ā–Ĺ–ĺ—Ā—ā–ł (70 200 —Ā–Ľ—É—á–į–Ķ–≤) [3].
–°–ĺ–≥–Ľ–į—Ā–Ĺ–ĺ –ī–į–Ĺ–Ĺ—č–ľ –ú–Ķ–∂–ī—É–Ĺ–į—Ä–ĺ–ī–Ĺ–ĺ–≥–ĺ –į–≥–Ķ–Ĺ—ā—Ā—ā–≤–į –Ņ–ĺ –ł–∑—É—á–Ķ–Ĺ–ł—é —Ä–į–ļ–į (International Agency for Research on Cancer, IARC), –†–ü–Ė –∑–į–Ĺ–ł–ľ–į–Ķ—ā 13-–Ķ –ľ–Ķ—Ā—ā–ĺ –Ņ–ĺ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ķ–ľ–ĺ—Ā—ā–ł –≤¬†–ľ–ł—Ä–Ķ, –į¬†–Ņ–ĺ —Ā–ľ–Ķ—Ä—ā–Ĺ–ĺ—Ā—ā–ł¬†‚Äď 8-–Ķ. –Ę–į–ļ, –≤¬†2009 –≥. –≤¬†–°–®–ź –†–ü–Ė –∑–į–Ī–ĺ–Ľ–Ķ–Ľ–ł 42 470 —á–Ķ–Ľ–ĺ–≤–Ķ–ļ (10-–Ķ –ľ–Ķ—Ā—ā–ĺ –ī–Ľ—Ź –ľ—É–∂—á–ł–Ĺ –ł¬†–∂–Ķ–Ĺ—Č–ł–Ĺ), –į¬†—É–ľ–Ķ—Ä–Ľ–ł 35 240, —Ā–ĺ–ĺ—ā–Ĺ–ĺ—ą–Ķ–Ĺ–ł–Ķ —Ā–ľ–Ķ—Ä—ā–Ĺ–ĺ—Ā—ā–ł –ł¬†–∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ķ–ľ–ĺ—Ā—ā–ł —Ā–ĺ—Ā—ā–į–≤–ł–Ľ–ĺ 0,83 [4].
–í —Ü–Ķ–Ľ–ĺ–ľ –Ņ—Ź—ā–ł–Ľ–Ķ—ā–Ĺ—Ź—Ź –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā—Ć –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö –†–ü–Ė –Ĺ–Ķ –ī–ĺ—Ā—ā–ł–≥–į–Ķ—ā 5%. –ü–ĺ—Ā–Ľ–Ķ —É—Ā—ā–į–Ĺ–ĺ–≤–Ľ–Ķ–Ĺ–ł—Ź –ī–ł–į–≥–Ĺ–ĺ–∑–į –ĺ–ī–ł–Ĺ –≥–ĺ–ī –Ņ–Ķ—Ä–Ķ–∂–ł–≤–į—é—ā 15,2% –ľ—É–∂—á–ł–Ĺ –ł¬†16,4% –∂–Ķ–Ĺ—Č–ł–Ĺ. –í¬†–°–®–ź –∑–į 25 –Ľ–Ķ—ā
(—Ā 1975 –Ņ–ĺ 2000 –≥.) –Ņ–ĺ–ļ–į–∑–į—ā–Ķ–Ľ—Ć –Ņ—Ź—ā–ł–Ľ–Ķ—ā–Ĺ–Ķ–Ļ –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā–ł –ī–Ľ—Ź –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā¬†–†–ü–Ė —É–≤–Ķ–Ľ–ł—á–ł–Ľ—Ā—Ź —Ā¬†3 –ī–ĺ 4%. –Ē–Ľ—Ź —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—Ź: –∑–į —ć—ā–ĺ—ā –∂–Ķ –Ņ–Ķ—Ä–ł–ĺ–ī –Ņ–ĺ–ļ–į–∑–į—ā–Ķ–Ľ—Ć –Ņ—Ź—ā–ł–Ľ–Ķ—ā–Ĺ–Ķ–Ļ –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā–ł –Ņ—Ä–ł —Ä–į–ļ–Ķ —ā–ĺ–Ľ—Ā—ā–ĺ–Ļ –ļ–ł—ą–ļ–ł –≤—č—Ä–ĺ—Ā –Ĺ–į 12%, –Ņ—Ä–ł —Ä–į–ļ–Ķ –Ņ–ł—Č–Ķ–≤–ĺ–ī–į¬†‚Äď –Ĺ–į 9%, —Ä–į–ļ–Ķ –∂–Ķ–Ľ—É–ī–ļ–į¬†‚Äď –Ĺ–į 8%.
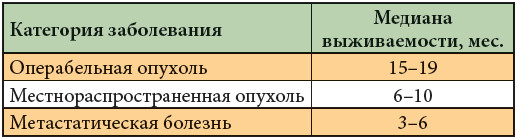
–õ–ł—ą—Ć 10% –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö –†–ü–Ė –Ņ–ĺ—ā–Ķ–Ĺ—Ü–ł–į–Ľ—Ć–Ĺ–ĺ –ĺ–Ņ–Ķ—Ä–į–Ī–Ķ–Ľ—Ć–Ĺ—č –Ĺ–į –ľ–ĺ–ľ–Ķ–Ĺ—ā –Ņ–Ķ—Ä–≤–ł—á–Ĺ–ĺ–Ļ –ī–ł–į–≥–Ĺ–ĺ—Ā—ā–ł–ļ–ł. –õ–ł–ľ—Ą–į—ā–ł—á–Ķ—Ā–ļ–į—Ź –ī–ł—Ā—Ā–Ķ–ľ–ł–Ĺ–į—Ü–ł—Ź –≤—č—Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –≤¬†45‚Äď70% —Ā–Ľ—É—á–į–Ķ–≤ –Ņ—Ä–ł –ĺ–Ņ—É—Ö–ĺ–Ľ—Ź—Ö —Ä–į–∑–ľ–Ķ—Ä–ĺ–ľ –ľ–Ķ–Ĺ–Ķ–Ķ 2 —Ā–ľ –≤¬†–ī–ł–į–ľ–Ķ—ā—Ä–Ķ. –Ē–ĺ 30% –≤–Ĺ–ĺ–≤—Ć –≤—č—Ź–≤–Ľ–Ķ–Ĺ–Ĺ—č—Ö –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö –ł–ľ–Ķ—é—ā –Ľ–ĺ–ļ–į–Ľ—Ć–Ĺ–ĺ —Ä–į—Ā–Ņ—Ä–ĺ—Ā—ā—Ä–į–Ĺ–Ķ–Ĺ–Ĺ—č–Ļ –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā, –į¬†60%¬†‚Äď –ĺ—ā–ī–į–Ľ–Ķ–Ĺ–Ĺ—č–Ķ –ľ–Ķ—ā–į—Ā—ā–į–∑—č [5]. –ú–Ķ–ī–ł–į–Ĺ—č –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā–ł –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö –≤¬†–∑–į–≤–ł—Ā–ł–ľ–ĺ—Ā—ā–ł –ĺ—ā —Ä–į—Ā–Ņ—Ä–ĺ—Ā—ā—Ä–į–Ĺ–Ķ–Ĺ–ł—Ź –ĺ–Ņ—É—Ö–ĺ–Ľ–ł –Ņ—Ä–Ķ–ī—Ā—ā–į–≤–Ľ–Ķ–Ĺ—č –≤¬†—ā–į–Ī–Ľ. 1.
–í –Ĺ–į—Ā—ā–ĺ—Ź—Č–Ķ–Ļ —Ā—ā–į—ā—Ć–Ķ –ľ—č –ĺ—Ā—ā–į–Ĺ–ĺ–≤–ł–ľ—Ā—Ź –Ĺ–į —Ö–ł–ľ–ł–ĺ—ā–Ķ—Ä–į–Ņ–ł–ł –ł¬†—ā–į—Ä–≥–Ķ—ā–Ĺ–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł –ī–ł—Ā—Ā–Ķ–ľ–ł–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–≥–ĺ –†–ü–Ė.
–•–ł–ľ–ł–ĺ—ā–Ķ—Ä–į–Ņ–ł—Ź –ī–ł—Ā—Ā–Ķ–ľ–ł–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–≥–ĺ –†–ü–Ė
–í–į–∂–Ĺ–ĺ–Ķ –∑–Ĺ–į—á–Ķ–Ĺ–ł–Ķ –ī–Ľ—Ź –Ľ–Ķ–ļ–į—Ä—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł –†–ü–Ė –ł–ľ–Ķ–Ľ–ĺ –ĺ—ā–ļ—Ä—č—ā–ł–Ķ –≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ–į. –ď–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ –Ī—č–Ľ –≤–ļ–Ľ—é—á–Ķ–Ĺ –≤¬†—Ā—ā–į–Ĺ–ī–į—Ä—ā–Ĺ—č–Ķ —Ā—Ö–Ķ–ľ—č –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź –ľ–Ķ—ā–į—Ā—ā–į—ā–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –†–ü–Ė –≤¬†1997 –≥., –Ņ–ĺ—Ā–Ľ–Ķ –Ņ—É–Ī–Ľ–ł–ļ–į—Ü–ł–ł —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā–ĺ–≤ —Ā–Ķ–≤–Ķ—Ä–ĺ–į–ľ–Ķ—Ä–ł–ļ–į–Ĺ—Ā–ļ–ĺ–≥–ĺ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź —Ā¬†—É—á–į—Ā—ā–ł–Ķ–ľ 126 –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤. –í¬†—ć—ā–ĺ–ľ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–ł II —Ą–į–∑—č –Ĺ–į —Ą–ĺ–Ĺ–Ķ –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź –≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ–ĺ–ľ –≤¬†—Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł–ł —Ā¬†5-—Ą—ā–ĺ—Ä—É—Ä–į—Ü–ł–Ľ–ĺ–ľ –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ķ —É–Ľ—É—á—ą–Ķ–Ĺ–ł–Ķ –ĺ—ā–ľ–Ķ—á–Ķ–Ĺ–ĺ —É¬†23,8 –ł¬†4,8% –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö, –ľ–Ķ–ī–ł–į–Ĺ–į –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā–ł —Ā–ĺ—Ā—ā–į–≤–ł–Ľ–į 5,65 –ł¬†4,41 –ľ–Ķ—Ā—Ź—Ü–į, –≥–ĺ–ī–ł—á–Ĺ–į—Ź –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā—Ƭ†‚Äď 18 –ł¬†2% —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ; —Ä–į–∑–Ĺ–ł—Ü–į –Ņ–ĺ –≤—Ā–Ķ–ľ –Ņ–ĺ–ļ–į–∑–į—ā–Ķ–Ľ—Ź–ľ –Ī—č–Ľ–į —Ā—ā–į—ā–ł—Ā—ā–ł—á–Ķ—Ā–ļ–ł –∑–Ĺ–į—á–ł–ľ–ĺ–Ļ; –Ņ–Ķ—Ä–Ķ–Ĺ–ĺ—Ā–ł–ľ–ĺ—Ā—ā—Ć –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź —É–ī–ĺ–≤–Ľ–Ķ—ā–≤–ĺ—Ä–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ļ [6]. –í¬†—ā–ĺ–ľ –∂–Ķ –≥–ĺ–ī—É A.M. Storniolo –ł¬†—Ā–ĺ–į–≤—ā. –ĺ–Ī–ĺ–Ī—Č–ł–Ľ–ł –ī–į–Ĺ–Ĺ—č–Ķ –嬆–ľ–ĺ–Ĺ–ĺ—ā–Ķ—Ä–į–Ņ–ł–ł –≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ–ĺ–ľ 3023 –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö –†–ü–Ė, –ľ–Ķ–ī–ł–į–Ĺ–į –Ņ—Ä–ĺ–ī–ĺ–Ľ–∂–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ—Ā—ā–ł –∂–ł–∑–Ĺ–ł –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā–ĺ—Ā—ā–į–≤–ł–Ľ–į 8 –ľ–Ķ—Ā—Ź—Ü–Ķ–≤ [7]. –ü—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł–Ķ –≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ–į –ī–ĺ–Ņ—É—Ā–ļ–į–Ķ—ā—Ā—Ź –ī–į–∂–Ķ –Ņ—Ä–ł –ĺ—Ü–Ķ–Ĺ–ļ–Ķ –ĺ–Ī—Č–Ķ–≥–ĺ —Ā–ĺ—Ā—ā–ĺ—Ź–Ĺ–ł—Ź –Ņ–į—Ü–ł–Ķ–Ĺ—ā–į –Ņ–ĺ —ą–ļ–į–Ľ–Ķ –ö–į—Ä–Ĺ–ĺ–≤—Ā–ļ–ĺ–≥–ĺ 60%.
–ü–ĺ—Ā–Ľ–Ķ –ļ–ĺ–Ĺ—Ā—ā–į—ā–į—Ü–ł–ł —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł –≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ–į –Ĺ–į—á–į–Ľ–ł—Ā—Ć —ą–ł—Ä–ĺ–ļ–ł–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź –Ķ–≥–ĺ –ļ–ĺ–ľ–Ī–ł–Ĺ–į—Ü–ł–Ļ —Ā¬†–ī—Ä—É–≥–ł–ľ–ł –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į–ľ–ł: 5-—Ą—ā–ĺ—Ä—É—Ä–į—Ü–ł–Ľ–ĺ–ľ, –ļ–į–Ņ–Ķ—Ü–ł—ā–į–Ī–ł–Ĺ–ĺ–ľ, UFT, S1, —ā–ĺ–ľ—É–ī–Ķ–ļ—Ā–ĺ–ľ, —Ü–ł—Ā–Ņ–Ľ–į—ā–ł–Ĺ–ĺ–ľ, –ĺ–ļ—Ā–į–Ľ–ł–Ņ–Ľ–į—ā–ł–Ĺ–ĺ–ľ, –ł—Ä–ł–Ĺ–ĺ—ā–Ķ–ļ–į–Ĺ–ĺ–ľ –ł¬†–ī—Ä. –í¬†—Ā–ĺ–≤—Ä–Ķ–ľ–Ķ–Ĺ–Ĺ–ĺ–Ļ –Ņ—Ä–į–ļ—ā–ł–ļ–Ķ —Ā—Ä–Ķ–ī–ł —Ö–ł–ľ–ł–ĺ—ā–Ķ—Ä–į–Ņ–Ķ–≤—ā–ł—á–Ķ—Ā–ļ–ł—Ö –ī—É–Ņ–Ľ–Ķ—ā–ĺ–≤ –Ĺ–į –ĺ—Ā–Ĺ–ĺ–≤–Ķ –≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ–į —á–į—Č–Ķ –ī—Ä—É–≥–ł—Ö –ł—Ā–Ņ–ĺ–Ľ—Ć–∑—É—é—ā –Ķ–≥–ĺ —Ā–ĺ—á–Ķ—ā–į–Ĺ–ł–Ķ —Ā¬†–Ņ—Ä–ĺ–ł–∑–≤–ĺ–ī–Ĺ—č–ľ–ł –Ņ–Ľ–į—ā–ł–Ĺ—č –ł¬†–ļ–į–Ņ–Ķ—Ü–ł—ā–į–Ī–ł–Ĺ–ĺ–ľ.
–í –Ķ–≤—Ä–ĺ–Ņ–Ķ–Ļ—Ā–ļ–ĺ–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ķ III —Ą–į–∑—č GERCOR / GISCAD –Ī—č–Ľ–ĺ —Ä–į–Ĺ–ī–ĺ–ľ–ł–∑–ł—Ä–ĺ–≤–į–Ĺ–ĺ 326 –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤, –ļ–ĺ—ā–ĺ—Ä—č–Ķ –Ņ–ĺ–Ľ—É—á–į–Ľ–ł –ļ–ĺ–ľ–Ī–ł–Ĺ–į—Ü–ł—é –≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ–į –ł¬†–ĺ–ļ—Ā–į–Ľ–ł–Ņ–Ľ–į—ā–ł–Ĺ–į –ł–Ľ–ł –ľ–ĺ–Ĺ–ĺ—ā–Ķ—Ä–į–Ņ–ł—é –≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ–ĺ–ľ. –í¬†–≥—Ä—É–Ņ–Ņ–Ķ, –Ņ–ĺ–Ľ—É—á–į–≤—ą–Ķ–Ļ —ā–Ķ—Ä–į–Ņ–ł—é –≤¬†—Ä–Ķ–∂–ł–ľ–Ķ GEMOX, –ĺ—ā–ľ–Ķ—á–Ķ–Ĺ—č –Ī–ĺ–Ľ–Ķ–Ķ –≤—č—Ā–ĺ–ļ–į—Ź —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź (26,8 –Ņ—Ä–ĺ—ā–ł–≤ 17,3%, —Ä = 0,04) –ł¬†–Ī–ĺ–Ľ–Ķ–Ķ –ī–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ–į—Ź –Ņ—Ä–ĺ–ī–ĺ–Ľ–∂–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ—Ā—ā—Ć –∂–ł–∑–Ĺ–ł –Ī–Ķ–∑ –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź (5,8 –Ņ—Ä–ĺ—ā–ł–≤ 3,7 –ľ–Ķ—Ā—Ź—Ü–į, —Ä = 0,04) –Ņ–ĺ —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—é —Ā¬†–Ņ–į—Ü–ł–Ķ–Ĺ—ā–į–ľ–ł –Ĺ–į –≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ–Ķ –≤¬†–ľ–ĺ–Ĺ–ĺ—Ä–Ķ–∂–ł–ľ–Ķ. –Ę–į–ļ–∂–Ķ –≤—č—Ź–≤–Ľ–Ķ–Ĺ–ĺ —Ā—ā–į—ā–ł—Ā—ā–ł—á–Ķ—Ā–ļ–ł –Ĺ–Ķ–∑–Ĺ–į—á–ł–ľ–ĺ–Ķ —É–≤–Ķ–Ľ–ł—á–Ķ–Ĺ–ł–Ķ –ĺ–Ī—Č–Ķ–Ļ –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā–ł: 9 –ł¬†7,1 –ľ–Ķ—Ā—Ź—Ü–į —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ (p = 0,13) [8].
–Ě–į –ĺ—Ā–Ĺ–ĺ–≤–į–Ĺ–ł–ł –Ĺ–Ķ—Ā–ļ–ĺ–Ľ—Ć–ļ–ł—Ö –ľ–Ķ—ā–į–į–Ĺ–į–Ľ–ł–∑–ĺ–≤, –Ņ—Ä–ĺ–≤–Ķ–ī–Ķ–Ĺ–Ĺ—č—Ö –ī–Ľ—Ź —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—Ź —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł –≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ–į –≤¬†–ľ–ĺ–Ĺ–ĺ—ā–Ķ—Ä–į–Ņ–ł–ł –ł¬†–≤ —Ā–ĺ—á–Ķ—ā–į–Ĺ–ł–ł —Ā¬†–Ņ—Ä–ĺ–ł–∑–≤–ĺ–ī–Ĺ—č–ľ–ł –Ņ–Ľ–į—ā–ł–Ĺ—č, –Ī—č–Ľ–ĺ –Ņ–ĺ–ļ–į–∑–į–Ĺ–ĺ –Ņ—Ä–Ķ–ł–ľ—É—Č–Ķ—Ā—ā–≤–ĺ –ļ–ĺ–ľ–Ī–ł–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł –≤¬†–ĺ—ā–Ĺ–ĺ—ą–Ķ–Ĺ–ł–ł —Ā—ā–į—ā–ł—Ā—ā–ł—á–Ķ—Ā–ļ–ł –∑–Ĺ–į—á–ł–ľ–ĺ–≥–ĺ –Ņ–ĺ–≤—č—ą–Ķ–Ĺ–ł—Ź —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź –ł¬†–ľ–Ķ–ī–ł–į–Ĺ—č –≤—Ä–Ķ–ľ–Ķ–Ĺ–ł –ī–ĺ –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź, –į¬†—ā–į–ļ–∂–Ķ —Ā–ļ—Ä–ĺ–ľ–Ĺ–ĺ–Ķ –ī–ĺ—Ā—ā–ĺ–≤–Ķ—Ä–Ĺ–ĺ–Ķ —É–≤–Ķ–Ľ–ł—á–Ķ–Ĺ–ł–Ķ –ĺ–Ī—Č–Ķ–Ļ –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā–ł [9‚Äď12].
–Ē—Ä—É–≥–ł–ľ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–ľ, –ļ–ĺ—ā–ĺ—Ä—č–Ļ —á–į—Č–Ķ –≤—Ā–Ķ–≥–ĺ –ļ–ĺ–ľ–Ī–ł–Ĺ–ł—Ä—É—é—ā —Ā¬†–≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ–ĺ–ľ, —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –ļ–į–Ņ–Ķ—Ü–ł—ā–į–Ī–ł–Ĺ. –°—Ä–į–≤–Ĺ–Ķ–Ĺ–ł–Ķ –ļ–ĺ–ľ–Ī–ł–Ĺ–į—Ü–ł–ł –≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ–į –ł¬†–ļ–į–Ņ–Ķ—Ü–ł—ā–į–Ī–ł–Ĺ–į —Ā¬†—Ä–Ķ–∂–ł–ľ–ĺ–ľ –ľ–ĺ–Ĺ–ĺ—ā–Ķ—Ä–į–Ņ–ł–ł –≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ–ĺ–ľ, –≤—č–Ņ–ĺ–Ľ–Ĺ–Ķ–Ĺ–Ĺ–ĺ–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į—ā–Ķ–Ľ—Ź–ľ–ł –ł–∑ –í–Ķ–Ľ–ł–ļ–ĺ–Ī—Ä–ł—ā–į–Ĺ–ł–ł, —Ā–≤–ł–ī–Ķ—ā–Ķ–Ľ—Ć—Ā—ā–≤–ĺ–≤–į–Ľ–ĺ –≤¬†–Ņ–ĺ–Ľ—Ć–∑—É –ļ–ĺ–ľ–Ī–ł–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł: —ć—Ą—Ą–Ķ–ļ—ā –Ņ–ĺ–Ľ—É—á–Ķ–Ĺ –≤¬†14,2% —Ā–Ľ—É—á–į–Ķ–≤ –ļ–ĺ–ľ–Ī–ł–Ĺ–į—Ü–ł–ł ¬ę–≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ + –ļ–į–Ņ–Ķ—Ü–ł—ā–į–Ī–ł–ŬĽ –ł¬†–≤ 7,1% —Ā–Ľ—É—á–į–Ķ–≤ –Ĺ–į –ĺ–ī–Ĺ–ĺ–ľ –≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ–Ķ (—Ä = 0,008), –ĺ–ī–Ĺ–ĺ–Ľ–Ķ—ā–Ĺ—Ź—Ź –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā—Ć —Ā–ĺ—Ā—ā–į–≤–ł–Ľ–į 23 –ł¬†17%
(—Ä = 0,023), –ľ–Ķ–ī–ł–į–Ĺ–į –ĺ–Ī—Č–Ķ–Ļ –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā–ł¬†‚Äď 7,4 –ł¬†6,0 –ľ–Ķ—Ā—Ź—Ü–į —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ (p > 0,05) [13].
–®–≤–Ķ–Ļ—Ü–į—Ä—Ā–ļ–į—Ź –≥—Ä—É–Ņ–Ņ–į –Ņ–ĺ –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł–ľ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź–ľ –ł¬†–≥—Ä—É–Ņ–Ņ–į ECOG (Eastern Cooperative Oncology Group¬†‚Äď –í–ĺ—Ā—ā–ĺ—á–Ĺ–į—Ź –ĺ–Ī—ä–Ķ–ī–ł–Ĺ–Ķ–Ĺ–Ĺ–į—Ź –≥—Ä—É–Ņ–Ņ–į –ĺ–Ĺ–ļ–ĺ–Ľ–ĺ–≥–ĺ–≤) —Ā—Ä–į–≤–Ĺ–ł–Ľ–ł –ľ–ĺ–Ĺ–ĺ—ā–Ķ—Ä–į–Ņ–ł—é –≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ–ĺ–ľ –ł¬†–ļ–ĺ–ľ–Ī–ł–Ĺ–į—Ü–ł—é –ļ–į–Ņ–Ķ—Ü–ł—ā–į–Ī–ł–Ĺ–į (650 –ľ–≥/–ľ2 –ī–≤–į —Ä–į–∑–į –≤¬†—Ā—É—ā–ļ–ł —Ā¬†1-–≥–ĺ –Ņ–ĺ 14-–Ļ –ī–Ķ–Ĺ—Ć) —Ā¬†–≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ–ĺ–ľ (1000 –ľ–≥/–ľ2 30-–ľ–ł–Ĺ—É—ā–Ĺ–į—Ź –ł–Ĺ—Ą—É–∑–ł—Ź –≤¬†1 –ł¬†8-–Ļ –ī–Ĺ–ł –ļ–į–∂–ī—č–Ķ 3 –Ĺ–Ķ–ī–Ķ–Ľ–ł) –≤¬†—Ä–į–Ĺ–ī–ĺ–ľ–ł–∑–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–ľ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–ł —Ā¬†—É—á–į—Ā—ā–ł–Ķ–ľ 319 –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö c —Ä–į—Ā–Ņ—Ä–ĺ—Ā—ā—Ä–į–Ĺ–Ķ–Ĺ–Ĺ—č–ľ –†–ü–Ė. –ú–Ķ–ī–ł–į–Ĺ–į –ĺ–Ī—Č–Ķ–Ļ –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā–ł —Ā–ĺ—Ā—ā–į–≤–ł–Ľ–į 8,4 –ł¬†7,2 –ľ–Ķ—Ā—Ź—Ü–į –≤¬†–Ņ–ĺ–Ľ—Ć–∑—É —Ä–Ķ–∂–ł–ľ–į GEMCAP, –Ĺ–ĺ –ī–į–Ĺ–Ĺ—č–Ķ –ĺ–ļ–į–∑–į–Ľ–ł—Ā—Ć —Ā—ā–į—ā–ł—Ā—ā–ł—á–Ķ—Ā–ļ–ł –Ĺ–Ķ–ī–ĺ—Ā—ā–ĺ–≤–Ķ—Ä–Ĺ—č–ľ–ł (—Ä = 0,234). –ě–ī–Ĺ–į–ļ–ĺ –ī–ĺ–Ņ–ĺ–Ľ–Ĺ–ł—ā–Ķ–Ľ—Ć–Ĺ—č–Ļ –į–Ĺ–į–Ľ–ł–∑ –≤¬†–Ņ–ĺ–ī–≥—Ä—É–Ņ–Ņ–Ķ –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤, –ĺ–Ī—Č–Ķ–Ķ —Ā–ĺ—Ā—ā–ĺ—Ź–Ĺ–ł–Ķ –ļ–ĺ—ā–ĺ—Ä—č—Ö –ĺ—Ü–Ķ–Ĺ–ł–≤–į–Ľ–ĺ—Ā—Ć –ļ–į–ļ ¬ę—Ö–ĺ—Ä–ĺ—ą–Ķ–Ķ¬Ľ (—Ā—ā–į—ā—É—Ā –Ņ–ĺ —ą–ļ–į–Ľ–Ķ –ö–į—Ä–Ĺ–ĺ–≤—Ā–ļ–ĺ–≥–ĺ –ĺ—ā 90 –ī–ĺ 100%), –Ņ—Ä–ĺ–ī–Ķ–ľ–ĺ–Ĺ—Ā—ā—Ä–ł—Ä–ĺ–≤–į–Ľ –∑–Ĺ–į—á–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ķ —É–≤–Ķ–Ľ–ł—á–Ķ–Ĺ–ł–Ķ –ľ–Ķ–ī–ł–į–Ĺ—č –ĺ–Ī—Č–Ķ–Ļ –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā–ł –Ņ—Ä–ł –ł—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į–Ĺ–ł–ł –ļ–ĺ–ľ–Ī–ł–Ĺ–į—Ü–ł–ł ¬ę–≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ + –ļ–į–Ņ–Ķ—Ü–ł—ā–į–Ī–ł–ŬĽ –Ņ–ĺ —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—é —Ā¬†–ľ–ĺ–Ĺ–ĺ—ā–Ķ—Ä–į–Ņ–ł–Ķ–Ļ –≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ–ĺ–ľ (10,1 –Ņ—Ä–ĺ—ā–ł–≤ 7,4 –ľ–Ķ—Ā—Ź—Ü–į —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ, —Ä = 0,014), –Ņ—Ä–ł —ć—ā–ĺ–ľ —á–į—Ā—ā–ĺ—ā–į –Ņ–ĺ–Ī–ĺ—á–Ĺ—č—Ö —ć—Ą—Ą–Ķ–ļ—ā–ĺ–≤ 3-–Ļ –ł–Ľ–ł 4-–Ļ —Ā—ā–Ķ–Ņ–Ķ–Ĺ–ł –Ī—č–Ľ–į –ĺ–ī–ł–Ĺ–į–ļ–ĺ–≤–ĺ–Ļ –≤¬†–ĺ–Ī–Ķ–ł—Ö –≥—Ä—É–Ņ–Ņ–į—Ö [14].
–Į–Ņ–ĺ–Ĺ—Ā–ļ–ł–ľ–ł –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į—ā–Ķ–Ľ—Ź–ľ–ł –ĺ–Ņ—É–Ī–Ľ–ł–ļ–ĺ–≤–į–Ĺ—č —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā—č –ľ–Ķ—ā–į–į–Ĺ–į–Ľ–ł–∑–į, –ĺ–Ī–ĺ–Ī—Č–į—é—Č–Ķ–≥–ĺ –ī–į–Ĺ–Ĺ—č–Ķ 18 —Ä–į–Ĺ–ī–ĺ–ľ–ł–∑–ł—Ä–ĺ–≤–į–Ĺ–Ĺ—č—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ, –ĺ–Ī—ä–Ķ–ī–ł–Ĺ–ł–≤—ą–ł—Ö 4237 –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā¬†—Ä–į—Ā–Ņ—Ä–ĺ—Ā—ā—Ä–į–Ĺ–Ķ–Ĺ–Ĺ—č–ľ –†–ü–Ė, –ļ–ĺ—ā–ĺ—Ä—č–Ķ –Ī—č–Ľ–ł —Ä–į–∑–ī–Ķ–Ľ–Ķ–Ĺ—č –Ĺ–į 5 –Ņ–ĺ–ī–≥—Ä—É–Ņ–Ņ –≤¬†–∑–į–≤–ł—Ā–ł–ľ–ĺ—Ā—ā–ł –ĺ—ā —Ā—Ö–Ķ–ľ—č –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź: ¬ę–≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ + –ļ–į–Ņ–Ķ—Ü–ł—ā–į–Ī–ł–ŬĽ, ¬ę–≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ + —Ü–ł—Ā–Ņ–Ľ–į—ā–ł–ŬĽ, ¬ę–≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ + 5-—Ą—ā–ĺ—Ä—É—Ä–į—Ü–ł–Ľ¬Ľ, ¬ę–≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ + –ł—Ä–ł–Ĺ–ĺ—ā–Ķ–ļ–į–ŬĽ –ł¬†¬ę–≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ + –ĺ–ļ—Ā–į–Ľ–ł–Ņ–Ľ–į—ā–ł–ŬĽ. –ź–Ĺ–į–Ľ–ł–∑ –ī–į–Ĺ–Ĺ—č—Ö –Ņ–ĺ –Ņ–į—Ä–į–ľ–Ķ—ā—Ä—É –ĺ–Ī—Č–Ķ–≥–ĺ —Ā–ĺ—Ā—ā–ĺ—Ź–Ĺ–ł—Ź –Ī–ĺ–Ľ—Ć–Ĺ–ĺ–≥–ĺ –≤—č–Ņ–ĺ–Ľ–Ĺ–Ķ–Ĺ –≤¬†4 –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź—Ö (1325 –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö). –í—č—Ź–≤–Ľ–Ķ–Ĺ–ĺ, —á—ā–ĺ –Ņ—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł–Ķ –ļ–ĺ–ľ–Ī–ł–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–Ļ —Ö–ł–ľ–ł–ĺ—ā–Ķ—Ä–į–Ņ–ł–ł —ɬ†–Ī–ĺ–Ľ—Ć–Ĺ—č—Ö —Ā¬†–Ņ–Ľ–ĺ—Ö–ł–ľ –ĺ–Ī—Č–ł–ľ —Ā–ĺ—Ā—ā–ĺ—Ź–Ĺ–ł–Ķ–ľ —É–≤–Ķ–Ľ–ł—á–ł–≤–į–Ķ—ā —Ä–ł—Ā–ļ —Ā–ľ–Ķ—Ä—ā–ł –≤¬†—ā–Ķ—á–Ķ–Ĺ–ł–Ķ 6 –ľ–Ķ—Ā—Ź—Ü–Ķ–≤ (1,17, —Ä = 0,04) –ł¬†–ĺ–ī–Ĺ–ĺ–≥–ĺ –≥–ĺ–ī–į (1,09, —Ä = 0,04) –ł, –Ĺ–į–Ņ—Ä–ĺ—ā–ł–≤, –ł—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į–Ĺ–ł–Ķ –ļ–ĺ–ľ–Ī–ł–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł —ɬ†–Ī–ĺ–Ľ—Ć–Ĺ—č—Ö —Ā¬†—Ö–ĺ—Ä–ĺ—ą–ł–ľ –ĺ–Ī—Č–ł–ľ —Ā–ĺ—Ā—ā–ĺ—Ź–Ĺ–ł–Ķ–ľ —Ā–Ĺ–ł–∂–į–Ķ—ā —Ä–ł—Ā–ļ —Ā–ľ–Ķ—Ä—ā–ł –≤¬†—ā–Ķ—á–Ķ–Ĺ–ł–Ķ –≥–ĺ–ī–į (0,93, —Ä = 0,08). –ú–Ķ—ā–į–į–Ĺ–į–Ľ–ł–∑ –Ņ—Ä–ĺ–ī–Ķ–ľ–ĺ–Ĺ—Ā—ā—Ä–ł—Ä–ĺ–≤–į–Ľ –∑–Ĺ–į—á–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ķ —É–≤–Ķ–Ľ–ł—á–Ķ–Ĺ–ł–Ķ –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā–ł –Ņ—Ä–ł —Ā–ĺ—á–Ķ—ā–į–Ĺ–ł–ł –≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ–į —Ā¬†–ļ–į–Ņ–Ķ—Ü–ł—ā–į–Ī–ł–Ĺ–ĺ–ľ –ł–Ľ–ł –ĺ–ļ—Ā–į–Ľ–ł–Ņ–Ľ–į—ā–ł–Ĺ–ĺ–ľ [15].
–í –ĺ—ā–Ķ—á–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ–Ļ –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ļ –Ņ—Ä–į–ļ—ā–ł–ļ–Ķ –≤—Ā–Ķ —Ä–Ķ–∂–Ķ –ł—Ā–Ņ–ĺ–Ľ—Ć–∑—É—é—ā –ĺ—Ä–ł–≥–ł–Ĺ–į–Ľ—Ć–Ĺ—č–Ķ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā—č, –∑–į–ľ–Ķ–Ĺ—Ź—Ź –ł—Ö –Ĺ–į –≤–ĺ—Ā–Ņ—Ä–ĺ–ł–∑–≤–ĺ–ī–ł–ľ—č–Ķ –≤¬†–†–ĺ—Ā—Ā–ł–Ļ—Ā–ļ–ĺ–Ļ –§–Ķ–ī–Ķ—Ä–į—Ü–ł–ł. –Ę–į–ļ, –ľ—č –Ņ—Ä–ł–ľ–Ķ–Ĺ—Ź–Ľ–ł –ĺ–ļ—Ā–į–Ľ–ł–Ņ–Ľ–į—ā–ł–Ĺ –ł¬†–≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ (–Ņ—Ä–Ķ–Ņ–į—Ä–į—ā—č –ļ–ĺ–ľ–Ņ–į–Ĺ–ł–ł –ě–ź–ě ¬ę–í–ē–†–ě–§–ź–†–ú¬Ľ) –≤¬†–ļ–į—á–Ķ—Ā—ā–≤–Ķ –ļ–ĺ–ľ–Ī–ł–Ĺ–į—Ü–ł–ł GEMOX –ī–Ľ—Ź –≤—ā–ĺ—Ä–ĺ–Ļ –Ľ–ł–Ĺ–ł–ł —ā–Ķ—Ä–į–Ņ–ł–ł –Ņ—Ä–ł –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł–ł –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź, –Ĺ–ĺ –Ĺ–Ķ —Ä–į–Ĺ—Ć—ą–Ķ —á–Ķ–ľ —á–Ķ—Ä–Ķ–∑ 3 –ľ–Ķ—Ā—Ź—Ü–į –Ņ–ĺ—Ā–Ľ–Ķ –Ņ—Ä–Ķ–ļ—Ä–į—Č–Ķ–Ĺ–ł—Ź —ā–Ķ—Ä–į–Ņ–ł–ł –≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ–ĺ–ľ (–Ņ—Ä–Ķ–Ņ–į—Ä–į—ā –ď–Ķ–ľ–∑–į—Ä). –í—Ā–Ķ–≥–ĺ –≤¬†–ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ķ –Ī—č–Ľ–ĺ –≤–ļ–Ľ—é—á–Ķ–Ĺ–ĺ 11 –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ (7 –∂–Ķ–Ĺ—Č–ł–Ĺ –ł¬†4 –ľ—É–∂—á–ł–Ĺ), –ľ–Ķ–ī–ł–į–Ĺ–į –≤–ĺ–∑—Ä–į—Ā—ā–į¬†‚Äď 68 –Ľ–Ķ—ā. –ě–Ī—Č–Ķ–Ķ —Ā–ĺ—Ā—ā–ĺ—Ź–Ĺ–ł–Ķ 8 –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö –ĺ—Ü–Ķ–Ĺ–ł–≤–į–Ľ–ĺ—Ā—Ć –ļ–į–ļ 1 –Ī–į–Ľ–Ľ –Ņ–ĺ —ą–ļ–į–Ľ–Ķ ECOG, —ā—Ä–ĺ–ł—Ö¬†‚Äď 2 –Ī–į–Ľ–Ľ–į. –í¬†—Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā–Ķ –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź –ĺ—ā–ľ–Ķ—á–Ķ–Ĺ—č –≤¬†–ĺ–ī–Ĺ–ĺ–ľ —Ā–Ľ—É—á–į–Ķ —á–į—Ā—ā–ł—á–Ĺ–į—Ź —Ä–Ķ–≥—Ä–Ķ—Ā—Ā–ł—Ź –ł¬†–≤ —á–Ķ—ā—č—Ä–Ķ—Ö¬†‚Äď —Ā—ā–į–Ī–ł–Ľ–ł–∑–į—Ü–ł—Ź
(—É –ī–≤—É—Ö –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤¬†‚Äď –Ī–ĺ–Ľ–Ķ–Ķ 5 –ľ–Ķ—Ā—Ź—Ü–Ķ–≤). –Ě–į –ĺ—Ā–Ĺ–ĺ–≤–į–Ĺ–ł–ł –Ņ–ĺ–Ľ—É—á–Ķ–Ĺ–Ĺ—č—Ö –ī–į–Ĺ–Ĺ—č—Ö –Ņ—Ä–Ķ–ī—Ā—ā–į–≤–Ľ—Ź–Ķ—ā—Ā—Ź –≤–ĺ–∑–ľ–ĺ–∂–Ĺ—č–ľ –≤–ĺ–∑–ĺ–Ī–Ĺ–ĺ–≤–Ľ–Ķ–Ĺ–ł–Ķ –≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ—Ā–ĺ–ī–Ķ—Ä–∂–į—Č–ł—Ö –ļ–ĺ–ľ–Ī–ł–Ĺ–į—Ü–ł–Ļ —ɬ†–Ī–ĺ–Ľ—Ć–Ĺ—č—Ö –≤¬†—Ö–ĺ—Ä–ĺ—ą–Ķ–ľ —Ā–ĺ—Ā—ā–ĺ—Ź–Ĺ–ł–ł –Ņ–ĺ—Ā–Ľ–Ķ –ľ–ĺ–Ĺ–ĺ—ā–Ķ—Ä–į–Ņ–ł–ł –ď–Ķ–ľ–∑–į—Ä–ĺ–ľ. –ü–ĺ–ī—á–Ķ—Ä–ļ–Ĺ–Ķ–ľ, —á—ā–ĺ –Ľ–Ķ—á–Ķ–Ĺ–ł–Ķ –ď–Ķ–ľ–∑–į—Ä–ĺ–ľ –ī–ĺ–Ľ–∂–Ĺ–ĺ –Ī—č—ā—Ć –Ņ—Ä–Ķ–ļ—Ä–į—Č–Ķ–Ĺ–ĺ –Ĺ–Ķ —Ä–į–Ĺ—Ć—ą–Ķ —á–Ķ–ľ –∑–į 3 –ľ–Ķ—Ā—Ź—Ü–į –ī–ĺ –Ĺ–į—á–į–Ľ–į —ā–Ķ—Ä–į–Ņ–ł–ł –≤—ā–ĺ—Ä–ĺ–Ļ –Ľ–ł–Ĺ–ł–ł. –ė–∑ –ĺ—Ā–ĺ–Ī–Ķ–Ĺ–Ĺ–ĺ—Ā—ā–Ķ–Ļ –Ņ—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł—Ź –ĺ—ā–Ķ—á–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ—č—Ö –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤ —Ā–Ľ–Ķ–ī—É–Ķ—ā –ĺ–Ī—Ä–į—ā–ł—ā—Ć –≤–Ĺ–ł–ľ–į–Ĺ–ł–Ķ –Ĺ–į –Ī–ĺ–Ľ–Ķ–Ķ —á–į—Ā—ā–ĺ–Ķ (—É –Ņ–ĺ–Ľ–ĺ–≤–ł–Ĺ—č –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö) —Ä–į–∑–≤–ł—ā–ł–Ķ –Ľ–ł—Ö–ĺ—Ä–į–ī–ļ–ł –ł¬†–į—Ā—ā–Ķ–Ĺ–ł–ł –Ņ–ĺ —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—é —Ā¬†–Ņ—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł–Ķ–ľ –ĺ—Ä–ł–≥–ł–Ĺ–į–Ľ—Ć–Ĺ—č—Ö –∑–į—Ä—É–Ī–Ķ–∂–Ĺ—č—Ö –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤.
–ě–ī–Ĺ–ł–ľ –ł–∑ –Ĺ–į–Ņ—Ä–į–≤–Ľ–Ķ–Ĺ–ł–Ļ –≤¬†–Ľ–Ķ—á–Ķ–Ĺ–ł–ł —Ä–į—Ā–Ņ—Ä–ĺ—Ā—ā—Ä–į–Ĺ–Ķ–Ĺ–Ĺ–ĺ–≥–ĺ –†–ü–Ė, –Ņ–ĺ–∑–≤–ĺ–Ľ–ł–≤—ą–ł—Ö —É–Ľ—É—á—ą–ł—ā—Ć —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā—č —ā–Ķ—Ä–į–Ņ–ł–ł, —Ā—ā–į–Ľ–ĺ –≤–≤–Ķ–ī–Ķ–Ĺ–ł–Ķ –≤¬†–Ņ—Ä–į–ļ—ā–ł–ļ—É —Ö–ł–ľ–ł–ĺ—ā–Ķ—Ä–į–Ņ–Ķ–≤—ā–ł—á–Ķ—Ā–ļ–ł—Ö —ā—Ä–ł–Ņ–Ľ–Ķ—ā–ĺ–≤. –í¬†2011 –≥. –Ī—č–Ľ–ł –ĺ–Ņ—É–Ī–Ľ–ł–ļ–ĺ–≤–į–Ĺ—č –ī–į–Ĺ–Ĺ—č–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź –ļ–ĺ–ľ–Ī–ł–Ĺ–į—Ü–ł–ł FOLFIRINOX (—Ą—ā–ĺ—Ä—É—Ä–į—Ü–ł–Ľ, –Ľ–Ķ–Ļ–ļ–ĺ–≤–ĺ—Ä–ł–Ĺ, –ł—Ä–ł–Ĺ–ĺ—ā–Ķ–ļ–į–Ĺ –ł¬†–ĺ–ļ—Ā–į–Ľ–ł–Ņ–Ľ–į—ā–ł–Ĺ) –≤¬†—Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł–ł —Ā¬†–ľ–ĺ–Ĺ–ĺ—ā–Ķ—Ä–į–Ņ–ł–Ķ–Ļ –≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ–ĺ–ľ. –£—Ā—ā–į–Ĺ–ĺ–≤–Ľ–Ķ–Ĺ–ĺ –ī–ĺ—Ā—ā–ĺ–≤–Ķ—Ä–Ĺ–ĺ–Ķ —É–≤–Ķ–Ľ–ł—á–Ķ–Ĺ–ł–Ķ –ľ–Ķ–ī–ł–į–Ĺ—č –ĺ–Ī—Č–Ķ–Ļ –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā–ł (11,1 –Ņ—Ä–ĺ—ā–ł–≤ 6,8 –ľ–Ķ—Ā—Ź—Ü–į, —Ä = 0,001), –ľ–Ķ–ī–ł–į–Ĺ—č –≤—Ä–Ķ–ľ–Ķ–Ĺ–ł –ī–ĺ –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź (6,4 –Ņ—Ä–ĺ—ā–ł–≤
3,3 –ľ–Ķ—Ā—Ź—Ü–į, —Ä = 0,001), —á–į—Ā—ā–ĺ—ā—č –ĺ–Ī—ä–Ķ–ļ—ā–ł–≤–Ĺ—č—Ö –ĺ—ā–≤–Ķ—ā–ĺ–≤ (33,6 –Ņ—Ä–ĺ—ā–ł–≤ 9,4%), —Ā–Ĺ–ł–∂–Ķ–Ĺ–ł–Ķ —Ä–ł—Ā–ļ–į —Ā–ľ–Ķ—Ä—ā–ł –Ĺ–į 47%. –ü—Ä–ł —ć—ā–ĺ–ľ —Ā–Ľ–Ķ–ī—É–Ķ—ā –ĺ—ā–ľ–Ķ—ā–ł—ā—Ć, —á—ā–ĺ –Ņ—Ä–ĺ–≤–Ķ–ī–Ķ–Ĺ–ł–Ķ –Ņ–ĺ–Ľ–ł—Ö–ł–ľ–ł–ĺ—ā–Ķ—Ä–į–Ņ–ł–ł —Ü–Ķ–Ľ–Ķ—Ā–ĺ–ĺ–Ī—Ä–į–∑–Ĺ–ĺ –ł¬†–≤–ĺ–∑–ľ–ĺ–∂–Ĺ–ĺ —É¬†–Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ –ľ–ĺ–Ľ–ĺ–∂–Ķ 65 –Ľ–Ķ—ā –≤¬†—Ö–ĺ—Ä–ĺ—ą–Ķ–ľ –ĺ–Ī—Č–Ķ–ľ —Ā–ĺ—Ā—ā–ĺ—Ź–Ĺ–ł–ł [16].
–ė–∑-–∑–į –Ĺ–ł–∑–ļ–ĺ–Ļ —á—É–≤—Ā—ā–≤–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ—Ā—ā–ł –į–ī–Ķ–Ĺ–ĺ–ļ–į—Ä—Ü–ł–Ĺ–ĺ–ľ—č –Ņ–ĺ–ī–∂–Ķ–Ľ—É–ī–ĺ—á–Ĺ–ĺ–Ļ –∂–Ķ–Ľ–Ķ–∑—č –ļ¬†–ļ–Ľ–į—Ā—Ā–ł—á–Ķ—Ā–ļ–ĺ–Ļ —Ö–ł–ľ–ł–ĺ—ā–Ķ—Ä–į–Ņ–ł–ł –≤¬†–ľ–ł—Ä–Ķ —ą–ł—Ä–ĺ–ļ–ĺ –ł–∑—É—á–į–Ķ—ā—Ā—Ź —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć —ā–į—Ä–≥–Ķ—ā–Ĺ—č—Ö –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤ –Ņ—Ä–ł –ī–į–Ĺ–Ĺ–ĺ–ľ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł–ł. –Ě–ł–∂–Ķ —Ä–į—Ā—Ā–ľ–ĺ—ā—Ä–Ķ–Ĺ—č –ĺ—Ā–Ĺ–ĺ–≤–Ĺ—č–Ķ –Ņ—É—ā–ł –Ņ–į—ā–ĺ–≥–Ķ–Ĺ–Ķ–∑–į –†–ü–Ė –ł¬†–ľ–ĺ–Ľ–Ķ–ļ—É–Ľ—Ź—Ä–Ĺ—č–Ķ –ľ–ł—ą–Ķ–Ĺ–ł –ī–Ľ—Ź —ā–į—Ä–≥–Ķ—ā–Ĺ–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł.
–Ę–į—Ä–≥–Ķ—ā–Ĺ–į—Ź —ā–Ķ—Ä–į–Ņ–ł—Ź –ī–ł—Ā—Ā–Ķ–ľ–ł–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–≥–ĺ –†–ü–Ė
–ú—É—ā–į—Ü–ł—Ź –≥–Ķ–Ĺ–į KRAS
–ü—Ä–ł –į–ī–Ķ–Ĺ–ĺ–ļ–į—Ä—Ü–ł–Ĺ–ĺ–ľ–Ķ –Ņ–ĺ–ī–∂–Ķ–Ľ—É–ī–ĺ—á–Ĺ–ĺ–Ļ –∂–Ķ–Ľ–Ķ–∑—č –ľ—É—ā–į—Ü–ł–ł –≥–Ķ–Ĺ–į KRAS –≤—Ā—ā—Ä–Ķ—á–į—é—ā—Ā—Ź, –Ņ–ĺ —Ä–į–∑–Ĺ—č–ľ –ī–į–Ĺ–Ĺ—č–ľ, –≤¬†74‚Äď100% —Ā–Ľ—É—á–į–Ķ–≤, –Ņ—Ä–ł —ć—ā–ĺ–ľ —á–į—Č–Ķ –≤—Ā–Ķ–≥–ĺ –Ľ–ĺ–ļ–į–Ľ–ł–∑—É—é—ā—Ā—Ź –≤¬†12, 13 –ł¬†67-–ľ –ļ–ĺ–ī–ĺ–Ĺ–į—Ö [17‚Äď20]. –≠—ā–ĺ—ā –Ņ—Ä–ĺ—ā–ĺ–ĺ–Ĺ–ļ–ĺ–≥–Ķ–Ĺ –ļ–ĺ–ī–ł—Ä—É–Ķ—ā –≤—č—Ā–ĺ–ļ–ĺ–≥–ĺ–ľ–ĺ–Ľ–ĺ–≥–ł—á–Ĺ—č–Ļ –ľ–Ķ–ľ–Ī—Ä–į–Ĺ–ĺ—Ā–≤—Ź–∑–į–Ĺ–Ĺ—č–Ļ –Ī–Ķ–Ľ–ĺ–ļ (p21ras) —Ā¬†–ľ–ĺ–Ľ–Ķ–ļ—É–Ľ—Ź—Ä–Ĺ–ĺ–Ļ –ľ–į—Ā—Ā–ĺ–Ļ 21 –ļ–Ē–į, –ļ–ĺ—ā–ĺ—Ä—č–Ļ —Ą—É–Ĺ–ļ—Ü–ł–ĺ–Ĺ–į–Ľ—Ć–Ĺ–ĺ –Ņ—Ä–Ķ–ī—Ā—ā–į–≤–Ľ—Ź–Ķ—ā —Ā–ĺ–Ī–ĺ–Ļ G-–Ī–Ķ–Ľ–ĺ–ļ. –°–≤—Ź–∑—č–≤–į—Ź –≥—É–į–Ĺ–ĺ–∑–ł–Ĺ—ā—Ä–ł—Ą–ĺ—Ā—Ą–į—ā (–ď–Ę–§), p21ras –Ņ–Ķ—Ä–Ķ—Ö–ĺ–ī–ł—ā –≤¬†–į–ļ—ā–ł–≤–Ĺ—É—é –ļ–ĺ–Ĺ—Ą–ĺ—Ä–ľ–į—Ü–ł—é, –ĺ—Ā—É—Č–Ķ—Ā—ā–≤–Ľ—Ź—é—Č—É—é –Ņ–Ķ—Ä–Ķ–ī–į—á—É —Ā–ł–≥–Ĺ–į–Ľ–į –ĺ—ā —Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–ĺ–≤, —Ä–į—Ā–Ņ–ĺ–Ľ–ĺ–∂–Ķ–Ĺ–Ĺ—č—Ö –Ĺ–į –Ņ–ĺ–≤–Ķ—Ä—Ö–Ĺ–ĺ—Ā—ā–ł –ļ–Ľ–Ķ—ā–ļ–ł, –ļ¬†—Ź–ī—Ä—É [21].
–Ę–ĺ—á–Ķ—á–Ĺ—č–Ķ –ľ—É—ā–į—Ü–ł–ł –≤¬†–≥–Ķ–Ĺ–Ķ –Ņ—Ä–ł–≤–ĺ–ī—Ź—ā –ļ¬†–∑–į–ľ–Ķ—Č–Ķ–Ĺ–ł—é –į–ľ–ł–Ĺ–ĺ–ļ–ł—Ā–Ľ–ĺ—ā–Ĺ—č—Ö –ĺ—Ā—ā–į—ā–ļ–ĺ–≤ –≤¬†–Ī–Ķ–Ľ–ļ–Ķ, —á—ā–ĺ –ĺ—Ā–Ľ–į–Ī–Ľ—Ź–Ķ—ā –ď–Ę–§-–į–∑–Ĺ—É—é –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć p21ras, –≤¬†—Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā–Ķ —á–Ķ–≥–ĺ —Ā–ĺ–∑–ī–į–Ķ—ā—Ā—Ź —ć—Ą—Ą–Ķ–ļ—ā –Ņ–ĺ—Ā—ā–ĺ—Ź–Ĺ–Ĺ–ĺ–≥–ĺ –Ņ—Ä–ĺ–≤–Ķ–ī–Ķ–Ĺ–ł—Ź –≤–Ĺ—É—ā—Ä–ł–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ–ĺ–≥–ĺ —Ā–ł–≥–Ĺ–į–Ľ–į [22].
–Ě–Ķ–ĺ–Ī—Ö–ĺ–ī–ł–ľ—č–ľ —É—Ā–Ľ–ĺ–≤–ł–Ķ–ľ –ī–Ľ—Ź —Ą—É–Ĺ–ļ—Ü–ł–ĺ–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź –Ī–Ķ–Ľ–ļ–į Ras —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –Ķ–≥–ĺ —Ā–≤—Ź–∑—Ć —Ā¬†–≤–Ĺ—É—ā—Ä–Ķ–Ĺ–Ĺ–Ķ–Ļ –Ņ–ĺ–≤–Ķ—Ä—Ö–Ĺ–ĺ—Ā—ā—Ć—é —Ü–ł—ā–ĺ–Ņ–Ľ–į–∑–ľ–į—ā–ł—á–Ķ—Ā–ļ–ĺ–Ļ –ľ–Ķ–ľ–Ī—Ä–į–Ĺ—č. –ü–ĺ—Ā–Ľ–Ķ —Ā–ł–Ĺ—ā–Ķ–∑–į –Ņ—Ä–ĺ-Ras –Ņ—Ä–ĺ—ā–Ķ–ł–Ĺ –≤¬†—Ü–ł—ā–ĺ–Ņ–Ľ–į–∑–ľ–Ķ –Ņ—Ä–Ķ—ā–Ķ—Ä–Ņ–Ķ–≤–į–Ķ—ā —Ä—Ź–ī –Ņ–ĺ—Ā—ā—ā—Ä–į–Ĺ—Ā–Ľ—Ź—Ü–ł–ĺ–Ĺ–Ĺ—č—Ö –ł–∑–ľ–Ķ–Ĺ–Ķ–Ĺ–ł–Ļ, –ĺ–ī–Ĺ–ł–ľ –ł–∑ –ļ–ĺ—ā–ĺ—Ä—č—Ö —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź —Ą–į—Ä–Ĺ–Ķ–∑–ł–Ľ–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ, –≤¬†—Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā–Ķ —á–Ķ–≥–ĺ –Ķ–≥–ĺ C-–ī–ĺ–ľ–Ķ–Ĺ —Ā—ā–į–Ĺ–ĺ–≤–ł—ā—Ā—Ź –≥–ł–ī—Ä–ĺ—Ą–ĺ–Ī–Ĺ—č–ľ –ł¬†–ĺ–Ī–Ķ—Ā–Ņ–Ķ—á–ł–≤–į–Ķ—ā –Ĺ–į–ī–Ķ–∂–Ĺ–ĺ–Ķ —Ā—Ü–Ķ–Ņ–Ľ–Ķ–Ĺ–ł–Ķ —Ā¬†–ľ–Ķ–ľ–Ī—Ä–į–Ĺ–ĺ–Ļ –ļ–Ľ–Ķ—ā–ļ–ł. –Ę–į–ļ–ł–ľ –ĺ–Ī—Ä–į–∑–ĺ–ľ, –ł–Ĺ–≥–ł–Ī–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ —Ą–Ķ—Ä–ľ–Ķ–Ĺ—ā–į —Ą–į—Ä–Ĺ–Ķ–∑–ł–Ľ—ā—Ä–į–Ĺ—Ā—Ą–Ķ—Ä–į–∑—č –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ¬†–ł–Ĺ–į–ļ—ā–ł–≤–į—Ü–ł–ł p21ras. –ö—Ä–ĺ–ľ–Ķ —ā–ĺ–≥–ĺ, —Ą–į—Ä–Ĺ–Ķ–∑–ł–Ľ–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ –Ĺ–Ķ–ĺ–Ī—Ö–ĺ–ī–ł–ľ–ĺ –ī–Ľ—Ź —Ą—É–Ĺ–ļ—Ü–ł–ĺ–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź –Ī–Ķ–Ľ–ļ–į RhoB –ł¬†–Ī–Ķ–Ľ–ļ–ĺ–≤ —Ā–ł–≥–Ĺ–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –Ņ—É—ā–ł PI3K/Akt2 [21].
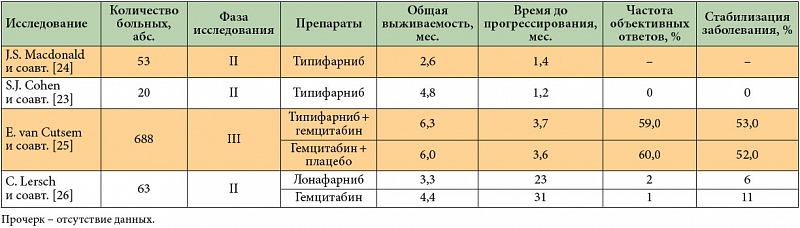
–Ę–ł–Ņ–ł—Ą–į—Ä–Ĺ–ł–Ī (R115777, –ó–į—Ä–Ĺ–Ķ—Ā—ā—Ä–į) —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź —Ā–Ķ–Ľ–Ķ–ļ—ā–ł–≤–Ĺ—č–ľ –ļ–ĺ–Ĺ–ļ—É—Ä–Ķ–Ĺ—ā–Ĺ—č–ľ –ł–Ĺ–≥–ł–Ī–ł—ā–ĺ—Ä–ĺ–ľ —Ą–į—Ä–Ĺ–Ķ–∑–ł–Ľ—ā—Ä–į–Ĺ—Ā—Ą–Ķ—Ä–į–∑—č. –Ě–Ķ—Ā–ľ–ĺ—ā—Ä—Ź –Ĺ–į –ľ–Ĺ–ĺ–≥–ĺ–ĺ–Ī–Ķ—Č–į—é—Č–ł–Ķ —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā—č –ľ–Ĺ–ĺ–≥–ł—Ö –ī–ĺ–ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö –ł—Ā–Ņ—č—ā–į–Ĺ–ł–Ļ, –Ņ—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł–Ķ —ā–ł–Ņ–ł—Ą–į—Ä–Ĺ–ł–Ī–į –ļ–į–ļ –≤¬†–ľ–ĺ–Ĺ–ĺ—ā–Ķ—Ä–į–Ņ–ł–ł, —ā–į–ļ –ł¬†–≤ –ļ–ĺ–ľ–Ī–ł–Ĺ–į—Ü–ł–ł —Ā¬†–≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ–ĺ–ľ –Ĺ–Ķ –Ņ—Ä–ł–≤–Ķ–Ľ–ĺ –ļ¬†—É–Ľ—É—á—ą–Ķ–Ĺ–ł—é —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā–ĺ–≤ –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź –†–ü–Ė –Ĺ–ł –≤¬†–ĺ–ī–Ĺ–ĺ–ľ –ł–∑ –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ II –ł¬†III —Ą–į–∑—č [23‚Äď25]. –°—Ö–ĺ–∂–ł–Ķ –ī–į–Ĺ–Ĺ—č–Ķ –Ī—č–Ľ–ł –Ņ–ĺ–Ľ—É—á–Ķ–Ĺ—č –ł¬†–≤ –ĺ—ā–Ĺ–ĺ—ą–Ķ–Ĺ–ł–ł –ł–Ĺ–≥–ł–Ī–ł—ā–ĺ—Ä–į —Ą–į—Ä–Ĺ–Ķ–∑–ł–Ľ—ā—Ä–į–Ĺ—Ā—Ą–Ķ—Ä–į–∑—č –≤—ā–ĺ—Ä–ĺ–≥–ĺ –Ņ–ĺ–ļ–ĺ–Ľ–Ķ–Ĺ–ł—Ź –Ľ–ĺ–Ĺ–į—Ą–į—Ä–Ĺ–ł–Ī–į (SCH66336) [26] (—ā–į–Ī–Ľ. 2).
MAPK-–ĺ–Ņ–ĺ—Ā—Ä–Ķ–ī–ĺ–≤–į–Ĺ–Ĺ—č–Ļ –≤–Ĺ—É—ā—Ä–ł–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—č–Ļ –Ņ—É—ā—Ć –Ņ–Ķ—Ä–Ķ–ī–į—á–ł —Ā–ł–≥–Ĺ–į–Ľ–į
–ü—Ä–ł —Ā–≤—Ź–∑—č–≤–į–Ĺ–ł–ł —Ä—Ź–ī–į —Ą–į–ļ—ā–ĺ—Ä–ĺ–≤ —Ä–ĺ—Ā—ā–į (EGF, PDGF), —Ü–ł—ā–ĺ–ļ–ł–Ĺ–ĺ–≤ (–ł–Ĺ—ā–Ķ—Ä–Ľ–Ķ–Ļ–ļ–ł–Ĺ–ĺ–≤-2,-3; G-CSF), –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–į —Ā¬†—ć–ļ—Ā—ā—Ä–į—Ü–Ķ–Ľ–Ľ—é–Ľ—Ź—Ä–Ĺ—č–ľ–ł –ī–ĺ–ľ–Ķ–Ĺ–į–ľ–ł —Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–ĺ–≤ –ļ¬†–Ĺ–ł–ľ –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–ī–ł—ā –į–ļ—ā–ł–≤–į—Ü–ł—Ź –Ī–Ķ–Ľ–ļ–į Ras, –ļ–ĺ—ā–ĺ—Ä—č–Ļ –≤¬†—Ā–≤–ĺ—é –ĺ—á–Ķ—Ä–Ķ–ī—Ć –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ¬†–∑–į–Ņ—É—Ā–ļ—É –Ĺ–Ķ—Ā–ļ–ĺ–Ľ—Ć–ļ–ł—Ö –≤–Ĺ—É—ā—Ä–ł–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—č—Ö —Ą–Ķ—Ä–ľ–Ķ–Ĺ—ā–į—ā–ł–≤–Ĺ—č—Ö –ļ–į—Ā–ļ–į–ī–ĺ–≤, –Ņ–Ķ—Ä–Ķ–ī–į—é—Č–ł—Ö —Ā–ł–≥–Ĺ–į–Ľ –ļ¬†–≥–Ķ–Ĺ–į–ľ, –ĺ—ā–≤–Ķ—ā—Ā—ā–≤–Ķ–Ĺ–Ĺ—č–ľ –∑–į –ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—É—é –Ņ—Ä–ĺ–Ľ–ł—Ą–Ķ—Ä–į—Ü–ł—é. –ě–ī–Ĺ–ł–ľ –ł–∑ —ā–į–ļ–ł—Ö —Ā–ł–≥–Ĺ–į–Ľ—Ć–Ĺ—č—Ö –Ņ—É—ā–Ķ–Ļ —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź Raf-1/MEK/MAPK/ERK/c-fos. –ö¬†–Ĺ–į—Ā—ā–ĺ—Ź—Č–Ķ–ľ—É –≤—Ä–Ķ–ľ–Ķ–Ĺ–ł —É–∂–Ķ —Ā–ł–Ĺ—ā–Ķ–∑–ł—Ä–ĺ–≤–į–Ĺ–ĺ –Ĺ–Ķ—Ā–ļ–ĺ–Ľ—Ć–ļ–ĺ –ľ–ĺ–Ľ–Ķ–ļ—É–Ľ, –Ī–Ľ–ĺ–ļ–ł—Ä—É—é—Č–ł—Ö –ľ–ł—ā–ĺ–≥–Ķ–Ĺ–į–ļ—ā–ł–≤–ł—Ä–ĺ–≤–į–Ĺ–Ĺ—č–Ķ –Ņ—Ä–ĺ—ā–Ķ–ł–Ĺ–ļ–ł–Ĺ–į–∑—č (AZD6244, CI-1040, PD0325901), –≤–Ķ–ī—É—ā—Ā—Ź –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź –Ņ–ĺ –ł–∑—É—á–Ķ–Ĺ–ł—é –ł—Ö —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł –Ņ—Ä–ł –†–ü–Ė [27]. –ě–Ņ—É–Ī–Ľ–ł–ļ–ĺ–≤–į–Ĺ—č —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā—č —ā–ĺ–Ľ—Ć–ļ–ĺ –ĺ–ī–Ĺ–ĺ–≥–ĺ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź II —Ą–į–∑—č, –≤¬†–ļ–ĺ—ā–ĺ—Ä–ĺ–ľ –ł–∑—É—á–į–Ľ–į—Ā—Ć —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į CI-1040 –Ņ—Ä–ł —Ä–į–∑–Ľ–ł—á–Ĺ—č—Ö —ā–ł–Ņ–į—Ö —Ā–ĺ–Ľ–ł–ī–Ĺ—č—Ö –ĺ–Ņ—É—Ö–ĺ–Ľ–Ķ–Ļ, –≤¬†—ā–ĺ–ľ —á–ł—Ā–Ľ–Ķ –ł¬†—Ā—Ä–Ķ–ī–ł –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā¬†–į–ī–Ķ–Ĺ–ĺ–ļ–į—Ä—Ü–ł–Ĺ–ĺ–ľ–ĺ–Ļ –Ņ–ĺ–ī–∂–Ķ–Ľ—É–ī–ĺ—á–Ĺ–ĺ–Ļ –∂–Ķ–Ľ–Ķ–∑—č. –ö–į–ļ–ł—Ö-–Ľ–ł–Ī–ĺ –∑–Ĺ–į—á–ł—ā–Ķ–Ľ—Ć–Ĺ—č—Ö —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā–ĺ–≤ –Ĺ–Ķ –Ņ–ĺ–Ľ—É—á–Ķ–Ĺ–ĺ: –Ĺ–ł —ɬ†–ĺ–ī–Ĺ–ĺ–≥–ĺ –ł–∑ –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ –Ĺ–Ķ –∑–į—Ä–Ķ–≥–ł—Ā—ā—Ä–ł—Ä–ĺ–≤–į–Ĺ–ĺ –ĺ–Ī—ä–Ķ–ļ—ā–ł–≤–Ĺ–ĺ–≥–ĺ –ĺ—ā–≤–Ķ—ā–į –Ĺ–į –Ľ–Ķ—á–Ķ–Ĺ–ł–Ķ, —ā–ĺ–Ľ—Ć–ļ–ĺ —É¬†2 (13%) –ł–∑ 15 –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö –†–ü–Ė —É–ī–į–Ľ–ĺ—Ā—Ć –ī–ĺ—Ā—ā–ł—á—Ć —Ā—ā–į–Ī–ł–Ľ–ł–∑–į—Ü–ł–ł –ĺ–Ņ—É—Ö–ĺ–Ľ–Ķ–≤–ĺ–≥–ĺ –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–į, –Ņ—Ä–ł —ć—ā–ĺ–ľ –≤—č—Ä–į–∂–Ķ–Ĺ–Ĺ—č—Ö –Ņ–ĺ–Ī–ĺ—á–Ĺ—č—Ö —ć—Ą—Ą–Ķ–ļ—ā–ĺ–≤ –Ĺ–Ķ –ĺ—ā–ľ–Ķ—á–Ķ–Ĺ–ĺ [28].
–ú—É–Ľ—Ć—ā–ł—ā–į—Ä–≥–Ķ—ā–Ĺ—č–ľ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–ľ, –ł–Ĺ–≥–ł–Ī–ł—Ä—É—é—Č–ł–ľ Raf-–ļ–ł–Ĺ–į–∑—č, —ā–ł—Ä–ĺ–∑–ł–Ĺ–ļ–ł–Ĺ–į–∑–Ĺ—č–Ķ –ī–ĺ–ľ–Ķ–Ĺ—č VEGFR-2, VEGFR-3 –ł¬†PDGFR-–Ī–Ķ—ā–į, —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź —Ā–ĺ—Ä–į—Ą–Ķ–Ĺ–ł–Ī. –í¬†–ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–ł I —Ą–į–∑—č –Ī—č–Ľ–į –Ņ—Ä–ĺ–ī–Ķ–ľ–ĺ–Ĺ—Ā—ā—Ä–ł—Ä–ĺ–≤–į–Ĺ–į —Ö–ĺ—Ä–ĺ—ą–į—Ź –Ņ–Ķ—Ä–Ķ–Ĺ–ĺ—Ā–ł–ľ–ĺ—Ā—ā—Ć –Ņ–į—Ü–ł–Ķ–Ĺ—ā–į–ľ–ł –ļ–ĺ–ľ–Ī–ł–Ĺ–į—Ü–ł–ł –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤ —Ā–ĺ—Ä–į—Ą–Ķ–Ĺ–ł–Ī –ł¬†–≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ [29]. –ě–ī–Ĺ–į–ļ–ĺ –≤¬†–ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–ľ –ł—Ā–Ņ—č—ā–į–Ĺ–ł–ł II —Ą–į–∑—č –Ĺ–Ķ –≤—č—Ź–≤–Ľ–Ķ–Ĺ–ĺ –Ķ–Ķ –Ņ—Ä–Ķ–ł–ľ—É—Č–Ķ—Ā—ā–≤ –Ņ–ĺ —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—é —Ā¬†—Ö–ł–ľ–ł–ĺ—ā–Ķ—Ä–į–Ņ–ł–Ķ–Ļ [30].
Src-–ĺ–Ņ–ĺ—Ā—Ä–Ķ–ī–ĺ–≤–į–Ĺ–Ĺ—č–Ļ –≤–Ĺ—É—ā—Ä–ł–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—č–Ļ –Ņ—É—ā—Ć –Ņ–Ķ—Ä–Ķ–ī–į—á–ł —Ā–ł–≥–Ĺ–į–Ľ–į
Src —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –ĺ–ī–Ĺ–ł–ľ –ł–∑ 9 –Ņ—Ä–Ķ–ī—Ā—ā–į–≤–ł—ā–Ķ–Ľ–Ķ–Ļ —Ā–Ķ–ľ–Ķ–Ļ—Ā—ā–≤–į –Ĺ–Ķ —Ā–≤—Ź–∑–į–Ĺ–Ĺ—č—Ö —Ā¬†—Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–ĺ–ľ —ā–ł—Ä–ĺ–∑–ł–Ĺ–ļ–ł–Ĺ–į–∑, –Ņ–ĺ–≤—č—ą–Ķ–Ĺ–Ĺ–į—Ź —ć–ļ—Ā–Ņ—Ä–Ķ—Ā—Ā–ł—Ź Src —Ö–į—Ä–į–ļ—ā–Ķ—Ä–Ĺ–į –ī–Ľ—Ź –ĺ–Ņ—É—Ö–ĺ–Ľ–Ķ–≤—č—Ö –ļ–Ľ–Ķ—ā–ĺ–ļ –†–ü–Ė [31]. Src –≤¬†–Ĺ–ĺ—Ä–ľ–Ķ¬†‚Äď –≤–į–∂–Ĺ–Ķ–Ļ—ą–ł–Ļ –ľ–Ķ–ī–ł–į—ā–ĺ—Ä –≤–Ĺ—É—ā—Ä–ł–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—č—Ö —Ā–ł–≥–Ĺ–į–Ľ—Ć–Ĺ—č—Ö –Ņ—É—ā–Ķ–Ļ, –į–ļ—ā–ł–≤–ł—Ä—É—é—Č–ł—Ö—Ā—Ź –Ņ—Ä–ł —Ā–≤—Ź–∑—č–≤–į–Ĺ–ł–ł —ć–Ņ–ł–ī–Ķ—Ä–ľ–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –ł¬†–ī—Ä—É–≥–ł—Ö —Ą–į–ļ—ā–ĺ—Ä–ĺ–≤ —Ä–ĺ—Ā—ā–į —Ā¬†—Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–į–ľ–ł. Src –į–ļ—ā–ł–≤–ł—Ä—É–Ķ—ā –ļ–ł–Ĺ–į–∑—É —Ą–ĺ–ļ–į–Ľ—Ć–Ĺ–ĺ–Ļ –į–ī–≥–Ķ–∑–ł–ł (FAK), —Ą–ĺ—Ā—Ą–ĺ—Ä–ł–Ľ–ł—Ä—É–Ķ—ā STAT-3 –ł¬†—Ā–≤—Ź–∑–į–Ĺ–Ĺ—č–Ķ —Ā¬†—Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–į–ľ–ł G-–Ī–Ķ–Ľ–ļ–ł, –≤–∑–į–ł–ľ–ĺ–ī–Ķ–Ļ—Ā—ā–≤—É–Ķ—ā —Ā¬†MAPK [32]. –ü–ĺ–≤—č—ą–Ķ–Ĺ–ł–Ķ –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł Src —Ā–Ņ–ĺ—Ā–ĺ–Ī—Ā—ā–≤—É–Ķ—ā –ľ–Ķ—ā–į—Ā—ā–į–∑–ł—Ä–ĺ–≤–į–Ĺ–ł—é –ĺ–Ņ—É—Ö–ĺ–Ľ–ł –∑–į —Ā—á–Ķ—ā —É—ā—Ä–į—ā—č –ļ–Ľ–Ķ—ā–ļ–į–ľ–ł –ľ–Ķ–∂–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—č—Ö –ļ–ĺ–Ĺ—ā–į–ļ—ā–ĺ–≤, –Ņ–Ķ—Ä–Ķ—Ā—ā—Ä–ĺ–Ļ–ļ–ł –į–ļ—ā–ł–Ĺ–ĺ–≤–ĺ–≥–ĺ —Ü–ł—ā–ĺ—Ā–ļ–Ķ–Ľ–Ķ—ā–į, –ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł—Ź —Ą–ĺ–ļ–į–Ľ—Ć–Ĺ—č—Ö –ļ–ĺ–Ĺ—ā–į–ļ—ā–ĺ–≤ [33].
–°–į—Ä–į–ļ–į—ā–ł–Ĺ–ł–Ī (AZD0530) —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –ľ–į–Ľ–ĺ–Ļ –ľ–ĺ–Ľ–Ķ–ļ—É–Ľ–ĺ–Ļ, –ł–Ĺ–≥–ł–Ī–ł—ā–ĺ—Ä–ĺ–ľ —ā–ł—Ä–ĺ–∑–ł–Ĺ–ļ–ł–Ĺ–į–∑ Src-—Ā–Ķ–ľ–Ķ–Ļ—Ā—ā–≤–į. –í¬†–ī–≤—É—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź—Ö II —Ą–į–∑—č, –≥–ī–Ķ —Ā–į—Ä–į–ļ–į—ā–ł–Ĺ–ł–Ī –Ņ—Ä–ł–ľ–Ķ–Ĺ—Ź–Ľ—Ā—Ź –ļ–į–ļ –≤¬†–ļ–ĺ–ľ–Ī–ł–Ĺ–į—Ü–ł–ł —Ā¬†–≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ–ĺ–ľ –≤¬†–ļ–į—á–Ķ—Ā—ā–≤–Ķ –Ņ–Ķ—Ä–≤–ĺ–Ļ –Ľ–ł–Ĺ–ł–ł —ā–Ķ—Ä–į–Ņ–ł–ł [34], —ā–į–ļ –ł¬†–≤ –ľ–ĺ–Ĺ–ĺ—ā–Ķ—Ä–į–Ņ–ł–ł –Ņ–ĺ—Ā–Ľ–Ķ –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź –Ĺ–į —Ą–ĺ–Ĺ–Ķ –≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ–į [35], –Ĺ–Ķ –Ņ–ĺ–Ľ—É—á–Ķ–Ĺ–ĺ –ļ–į–ļ–ł—Ö-–Ľ–ł–Ī–ĺ –ī–į–Ĺ–Ĺ—č—Ö –ĺ–Ī —É–Ľ—É—á—ą–Ķ–Ĺ–ł–ł —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā–ĺ–≤ –Ņ–ĺ —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—é —Ā–ĺ —Ā—ā–į–Ĺ–ī–į—Ä—ā–Ĺ—č–ľ–ł —Ā—Ö–Ķ–ľ–į–ľ–ł —Ö–ł–ľ–ł–ĺ—ā–Ķ—Ä–į–Ņ–ł–ł.
–í –Ĺ–į—Ā—ā–ĺ—Ź—Č–Ķ–Ķ –≤—Ä–Ķ–ľ—Ź –ł–ī–Ķ—ā –Ĺ–į–Ī–ĺ—Ä –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ –≤¬†–ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ķ II —Ą–į–∑—č –Ņ–ĺ –ł–∑—É—á–Ķ–Ĺ–ł—é –≤–Ľ–ł—Ź–Ĺ–ł—Ź –ł–Ĺ–≥–ł–Ī–ł—ā–ĺ—Ä–į Src-–ļ–ł–Ĺ–į–∑ –≤—ā–ĺ—Ä–ĺ–≥–ĺ –Ņ–ĺ–ļ–ĺ–Ľ–Ķ–Ĺ–ł—Ź –ī–į–∑–į—ā–ł–Ĺ–ł–Ī–į –ļ–į–ļ —Ą–į–ļ—ā–ĺ—Ä–į, –Ņ—Ä–Ķ–Ņ—Ź—ā—Ā—ā–≤—É—é—Č–Ķ–≥–ĺ –ī–ł—Ā—Ā–Ķ–ľ–ł–Ĺ–į—Ü–ł–ł –Ņ—Ä–ł –ľ–Ķ—Ā—ā–Ĺ–ĺ—Ä–į—Ā–Ņ—Ä–ĺ—Ā—ā—Ä–į–Ĺ–Ķ–Ĺ–Ĺ–ĺ–ľ –†–ü–Ė [36].
–ė–Ĺ–≥–ł–Ī–ł—ā–ĺ—Ä—č –ľ–į—ā—Ä–ł–ļ—Ā–Ĺ—č—Ö –ľ–Ķ—ā–į–Ľ–Ľ–ĺ–Ņ—Ä–ĺ—ā–Ķ–ł–Ĺ–į–∑
–ē—Č–Ķ –ĺ–ī–Ĺ–ł–ľ –Ĺ–į–Ņ—Ä–į–≤–Ľ–Ķ–Ĺ–ł–Ķ–ľ –≤¬†–Ľ–Ķ—á–Ķ–Ĺ–ł–ł –†–ü–Ė –Ī—č–Ľ–ĺ –≤–ļ–Ľ—é—á–Ķ–Ĺ–ł–Ķ –≤¬†–Ņ—Ä–ĺ—ā–ł–≤–ĺ–ĺ–Ņ—É—Ö–ĺ–Ľ–Ķ–≤—č–Ķ –ļ–ĺ–ľ–Ī–ł–Ĺ–į—Ü–ł–ł –ł–Ĺ–≥–ł–Ī–ł—ā–ĺ—Ä–ĺ–≤ –ľ–į—ā—Ä–ł–ļ—Ā–Ĺ—č—Ö –ľ–Ķ—ā–į–Ľ–Ľ–ĺ–Ņ—Ä–ĺ—ā–Ķ–ł–Ĺ–į–∑ (MMPs). MMPs –Ņ—Ä–Ķ–ī—Ā—ā–į–≤–Ľ—Ź—é—ā —Ā–ĺ–Ī–ĺ–Ļ —Ā–Ķ–ľ–Ķ–Ļ—Ā—ā–≤–ĺ —Ü–ł–Ĺ–ļ- –ł¬†–ļ–į–Ľ—Ć—Ü–ł–Ļ–∑–į–≤–ł—Ā–ł–ľ—č—Ö —ć–Ĺ–ī–ĺ–Ņ–Ķ–Ņ—ā–ł–ī–į–∑, –ļ–ĺ—ā–ĺ—Ä—č–Ķ –∑–į —Ā—á–Ķ—ā –ī–Ķ–≥—Ä–į–ī–į—Ü–ł–ł —ć–ļ—Ā—ā—Ä–į—Ü–Ķ–Ľ–Ľ—é–Ľ—Ź—Ä–Ĺ–ĺ–≥–ĺ –ľ–į—ā—Ä–ł–ļ—Ā–į –ł¬†—Ā—ā—Ä–ĺ–ľ—č —Ā–Ņ–ĺ—Ā–ĺ–Ī—Ā—ā–≤—É—é—ā –ľ–ł–≥—Ä–į—Ü–ł–ł –∑–Ľ–ĺ–ļ–į—á–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ—č—Ö –ļ–Ľ–Ķ—ā–ĺ–ļ. –°–Ķ–ľ–Ķ–Ļ—Ā—ā–≤–ĺ –ľ–į—ā—Ä–ł–ļ—Ā–Ĺ—č—Ö –ľ–Ķ—ā–į–Ľ–Ľ–ĺ–Ņ—Ä–ĺ—ā–Ķ–ł–Ĺ–į–∑ —Ā–ĺ—Ā—ā–ĺ–ł—ā –ł–∑ 20 —Ą–Ķ—Ä–ľ–Ķ–Ĺ—ā–ĺ–≤, —Ā–Ņ–ĺ—Ā–ĺ–Ī–Ĺ—č—Ö —Ä–į—Ā—Č–Ķ–Ņ–Ľ—Ź—ā—Ć –Ņ—Ä–į–ļ—ā–ł—á–Ķ—Ā–ļ–ł –≤—Ā–Ķ –ļ–ĺ–ľ–Ņ–ĺ–Ĺ–Ķ–Ĺ—ā—č –≤–Ĺ–Ķ–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ–ĺ–≥–ĺ –ľ–į—ā—Ä–ł–ļ—Ā–į.
–£—Ä–ĺ–≤–Ķ–Ĺ—Ć —ć–ļ—Ā–Ņ—Ä–Ķ—Ā—Ā–ł–ł MPPs –≤¬†–ĺ–Ņ—É—Ö–ĺ–Ľ–ł –∑–Ĺ–į—á–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ –≤—č—ą–Ķ, —á–Ķ–ľ –≤¬†–Ĺ–ĺ—Ä–ľ–į–Ľ—Ć–Ĺ–ĺ–Ļ —ā–ļ–į–Ĺ–ł –Ņ–ĺ–ī–∂–Ķ–Ľ—É–ī–ĺ—á–Ĺ–ĺ–Ļ –∂–Ķ–Ľ–Ķ–∑—č. –ě—Ā–Ĺ–ĺ–≤–Ĺ—č–ľ–ł –Ņ—Ä–ĺ—ā–Ķ–ł–Ĺ–į–∑–į–ľ–ł, –≤–ĺ–≤–Ľ–Ķ—á–Ķ–Ĺ–Ĺ—č–ľ–ł –≤¬†–Ņ–į—ā–ĺ–≥–Ķ–Ĺ–Ķ–∑ –†–ü–Ė, —Ź–≤–Ľ—Ź—é—ā—Ā—Ź MMP-2 –ł¬†MMP-9 [37]. –í—č—Ā–ĺ–ļ–ł–Ļ —É—Ä–ĺ–≤–Ķ–Ĺ—Ć —ć–ļ—Ā–Ņ—Ä–Ķ—Ā—Ā–ł–ł MMP-2 –ļ–ĺ—Ä—Ä–Ķ–Ľ–ł—Ä—É–Ķ—ā —Ā¬†–Ņ–Ľ–ĺ—Ö–ł–ľ –Ņ—Ä–ĺ–≥–Ĺ–ĺ–∑–ĺ–ľ –ł¬†–Ī—č—Ā—ā—Ä—č–ľ –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ–ľ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź [38].
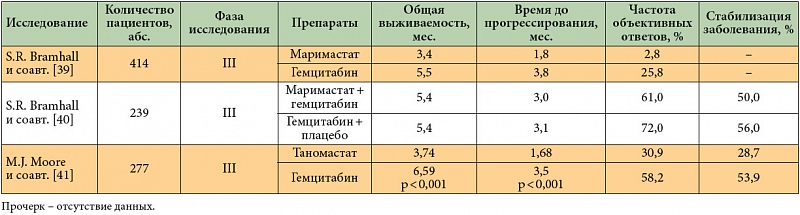
–ú–į—Ä–ł–ľ–į—Ā—ā–į—ā¬†‚Äď –Ņ–Ķ—Ä–≤—č–Ļ –ł¬†–Ĺ–į–ł–Ī–ĺ–Ľ–Ķ–Ķ –ł–∑—É—á–Ķ–Ĺ–Ĺ—č–Ļ –Ņ—Ä–ł —Ā–ĺ–Ľ–ł–ī–Ĺ—č—Ö –ĺ–Ņ—É—Ö–ĺ–Ľ—Ź—Ö –ł–Ĺ–≥–ł–Ī–ł—ā–ĺ—Ä MMPs 1, 2, 3, 7 –ł¬†9-–≥–ĺ —ā–ł–Ņ–į. –ě–ī–Ĺ–į–ļ–ĺ –≤¬†–ī–≤—É—Ö —Ä–į–Ĺ–ī–ĺ–ľ–ł–∑–ł—Ä–ĺ–≤–į–Ĺ–Ĺ—č—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź—Ö III —Ą–į–∑—č –Ņ—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł–Ķ –ľ–į—Ä–ł–ľ–į—Ā—ā–į—ā–į –≤¬†–ľ–ĺ–Ĺ–ĺ—ā–Ķ—Ä–į–Ņ–ł–ł –ł–Ľ–ł –≤¬†–ļ–ĺ–ľ–Ī–ł–Ĺ–į—Ü–ł–ł —Ā¬†–≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ–ĺ–ľ –Ĺ–Ķ –Ņ–ĺ–ļ–į–∑–į–Ľ–ĺ –Ĺ–ł–ļ–į–ļ–ł—Ö –Ņ—Ä–Ķ–ł–ľ—É—Č–Ķ—Ā—ā–≤ –Ņ–Ķ—Ä–Ķ–ī —Ā—ā–į–Ĺ–ī–į—Ä—ā–Ĺ–ĺ–Ļ —Ö–ł–ľ–ł–ĺ—ā–Ķ—Ä–į–Ņ–ł–Ķ–Ļ –†–ü–Ė [39, 40]. –ö—Ä–ĺ–ľ–Ķ —ā–ĺ–≥–ĺ, —ā–į—Ä–≥–Ķ—ā–Ĺ—č–Ļ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā –ī–į–Ķ—ā —Ā–Ņ–Ķ—Ü–ł—Ą–ł—á–Ķ—Ā–ļ–ł–Ķ –Ņ–ĺ–Ī–ĺ—á–Ĺ—č–Ķ —ć—Ą—Ą–Ķ–ļ—ā—č (–į—Ä—ā—Ä–į–Ľ–≥–ł–ł, –ľ—č—ą–Ķ—á–Ĺ–ĺ-–ļ–ĺ—Ā—ā–Ĺ—č–Ķ –Ī–ĺ–Ľ–ł, –Ņ–ĺ—Ź–≤–Ľ–Ķ–Ĺ–ł–Ķ —Ā–ļ–ĺ–≤–į–Ĺ–Ĺ–ĺ—Ā—ā–ł –ī–≤–ł–∂–Ķ–Ĺ–ł–Ļ).
–Ě–į–Ī–ĺ—Ä –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ –≤¬†–ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ķ III —Ą–į–∑—č –Ņ–ĺ –ł–∑—É—á–Ķ–Ĺ–ł—é –ł–Ĺ–≥–ł–Ī–ł—ā–ĺ—Ä–į MMPs –≤—ā–ĺ—Ä–ĺ–≥–ĺ –Ņ–ĺ–ļ–ĺ–Ľ–Ķ–Ĺ–ł—Ź —ā–į–Ĺ–ĺ–ľ–į—Ā—ā–į—ā–į (BAY 12-9566) –Ī—č–Ľ –ī–ĺ—Ā—Ä–ĺ—á–Ĺ–ĺ –∑–į–≤–Ķ—Ä—ą–Ķ–Ĺ –≤¬†—Ā–≤—Ź–∑–ł —Ā¬†—ā–Ķ–ľ, —á—ā–ĺ –Ņ—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł–Ķ –ī–į–Ĺ–Ĺ–ĺ–≥–ĺ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į –Ņ—Ä–ł–≤–ĺ–ī–ł–Ľ–ĺ –ļ¬†—Ā—ā–į—ā–ł—Ā—ā–ł—á–Ķ—Ā–ļ–ł –∑–Ĺ–į—á–ł–ľ–ĺ–ľ—É —Ā–ĺ–ļ—Ä–į—Č–Ķ–Ĺ–ł—é –Ņ—Ä–ĺ–ī–ĺ–Ľ–∂–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ—Ā—ā–ł –∂–ł–∑–Ĺ–ł –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ [41].
–°–Ķ–Ľ–Ķ–ļ—ā–ł–≤–Ĺ—č–Ļ –ł–Ĺ–≥–ł–Ī–ł—ā–ĺ—Ä –ľ–Ķ—ā–į–Ľ–Ľ–ĺ–Ņ—Ä–ĺ—ā–Ķ–ł–Ĺ–į–∑ 2 –ł¬†9-–≥–ĺ —ā–ł–Ņ–į Ro 28-2653 –Ņ—Ä–ĺ—Ö–ĺ–ī–ł—ā –≤¬†–Ĺ–į—Ā—ā–ĺ—Ź—Č–Ķ–Ķ –≤—Ä–Ķ–ľ—Ź –ī–ĺ–ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł–Ķ –ł—Ā–Ņ—č—ā–į–Ĺ–ł—Ź [42]. –†–Ķ–∑—É–Ľ—Ć—ā–į—ā—č –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ –ł–Ĺ–≥–ł–Ī–ł—ā–ĺ—Ä–ĺ–≤ MMPs –Ņ—Ä–ł –†–ü–Ė –ĺ–Ī–ĺ–Ī—Č–Ķ–Ĺ—č –≤¬†—ā–į–Ī–Ľ. 3.
–†–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä—č —ć–Ņ–ł–ī–Ķ—Ä–ľ–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ —Ą–į–ļ—ā–ĺ—Ä–į —Ä–ĺ—Ā—ā–į
–°–Ķ–ľ–Ķ–Ļ—Ā—ā–≤–ĺ —Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–ĺ–≤ —ć–Ņ–ł–ī–Ķ—Ä–ľ–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ —Ą–į–ļ—ā–ĺ—Ä–į —Ä–ĺ—Ā—ā–į (EGFR) –Ņ—Ä–Ķ–ī—Ā—ā–į–≤–Ľ—Ź–Ķ—ā —Ā–ĺ–Ī–ĺ–Ļ –≥—Ä—É–Ņ–Ņ—É —ā—Ä–į–Ĺ—Ā–ľ–Ķ–ľ–Ī—Ä–į–Ĺ–Ĺ—č—Ö –Ī–Ķ–Ľ–ļ–ĺ–≤, –ĺ—ā–≤–Ķ—ā—Ā—ā–≤–Ķ–Ĺ–Ĺ—č—Ö –∑–į –ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—É—é –Ņ—Ä–ĺ–Ľ–ł—Ą–Ķ—Ä–į—Ü–ł—é. –ė–∑–≤–Ķ—Ā—ā–Ĺ–ĺ 4 –≤–ł–ī–į —ā–į–ļ–ł—Ö —Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–ĺ–≤: EGFR (HER1, –ł–Ľ–ł ErbB1), HER2/neu (ErbB2), HER3 (ErbB3) –ł¬†HER4 (ErbB4). –ü–ĺ–≤—č—ą–Ķ–Ĺ–Ĺ–į—Ź —ć–ļ—Ā–Ņ—Ä–Ķ—Ā—Ā–ł—Ź EGFR –ł¬†–Ķ–≥–ĺ –Ľ–ł–≥–į–Ĺ–ī–ĺ–≤ EGF –ł¬†TGF-–į–Ľ—Ć—Ą–į —Ö–į—Ä–į–ļ—ā–Ķ—Ä–Ĺ–į –ī–Ľ—Ź –†–ü–Ė [43]. –í—č—Ā–ĺ–ļ–ł–Ļ —É—Ä–ĺ–≤–Ķ–Ĺ—Ć EGFR –≤¬†–ĺ–Ņ—É—Ö–ĺ–Ľ–ł —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –Ĺ–Ķ–Ī–Ľ–į–≥–ĺ–Ņ—Ä–ł—Ź—ā–Ĺ—č–ľ –Ņ—Ä–ĺ–≥–Ĺ–ĺ—Ā—ā–ł—á–Ķ—Ā–ļ–ł–ľ —Ą–į–ļ—ā–ĺ—Ä–ĺ–ľ, —Ā–≤—Ź–∑–į–Ĺ —Ā¬†–Ī–ĺ–Ľ–Ķ–Ķ –į–≥—Ä–Ķ—Ā—Ā–ł–≤–Ĺ—č–ľ —ā–Ķ—á–Ķ–Ĺ–ł–Ķ–ľ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź, —Ä–Ķ–∑–ł—Ā—ā–Ķ–Ĺ—ā–Ĺ–ĺ—Ā—ā—Ć—é –ļ¬†–Ľ—É—á–Ķ–≤–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł [44]. –°–Ķ–Ļ—á–į—Ā –≤¬†–ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ļ –Ņ—Ä–į–ļ—ā–ł–ļ–Ķ –Ņ—Ä–ł–ľ–Ķ–Ĺ—Ź—é—ā—Ā—Ź –ī–≤–Ķ –≥—Ä—É–Ņ–Ņ—č —ā–į—Ä–≥–Ķ—ā–Ĺ—č—Ö –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤, –Ī–Ľ–ĺ–ļ–ł—Ä—É—é—Č–ł—Ö EGFR-–Ņ—É—ā—Ć: –ľ–ĺ–Ĺ–ĺ–ļ–Ľ–ĺ–Ĺ–į–Ľ—Ć–Ĺ—č–Ķ –į–Ĺ—ā–ł—ā–Ķ–Ľ–į –Ĺ–Ķ–Ņ–ĺ—Ā—Ä–Ķ–ī—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ –ļ¬†–≤–Ĺ–Ķ–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ–ĺ–ľ—É –ī–ĺ–ľ–Ķ–Ĺ—É —Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–į (—Ü–Ķ—ā—É–ļ—Ā–ł–ľ–į–Ī, –Ņ–į–Ĺ–ł—ā—É–ľ—É–ľ–į–Ī) –ł¬†–ľ–į–Ľ—č–Ķ –ľ–ĺ–Ľ–Ķ–ļ—É–Ľ—č, –Ī–Ľ–ĺ–ļ–ł—Ä—É—é—Č–ł–Ķ –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć —ā–ł—Ä–ĺ–∑–ł–Ĺ–ļ–ł–Ĺ–į–∑–Ĺ–ĺ–≥–ĺ –ī–ĺ–ľ–Ķ–Ĺ–į —Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–į (–≥–Ķ—Ą–ł—ā–ł–Ĺ–ł–Ī, —ć—Ä–Ľ–ĺ—ā–ł–Ĺ–ł–Ī).
–≠—Ä–Ľ–ĺ—ā–ł–Ĺ–ł–Ī –Ĺ–į —Ā–Ķ–≥–ĺ–ī–Ĺ—Ź—ą–Ĺ–ł–Ļ –ī–Ķ–ŗƬ†‚Äď –Ķ–ī–ł–Ĺ—Ā—ā–≤–Ķ–Ĺ–Ĺ—č–Ļ —ā–į—Ä–≥–Ķ—ā–Ĺ—č–Ļ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā, –ĺ–ī–ĺ–Ī—Ä–Ķ–Ĺ–Ĺ—č–Ļ –£–Ņ—Ä–į–≤–Ľ–Ķ–Ĺ–ł–Ķ–ľ –Ņ–ĺ –ļ–ĺ–Ĺ—ā—Ä–ĺ–Ľ—é –Ņ–ł—Č–Ķ–≤—č—Ö –Ņ—Ä–ĺ–ī—É–ļ—ā–ĺ–≤ –ł¬†–Ľ–Ķ–ļ–į—Ä—Ā—ā–≤–Ķ–Ĺ–Ĺ—č—Ö –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤ –°–®–ź (Food and Drug Administration, FDA) –ī–Ľ—Ź –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö –†–ü–Ė. –í¬†2007 –≥. –Ī—č–Ľ–ł –ĺ–Ņ—É–Ī–Ľ–ł–ļ–ĺ–≤–į–Ĺ—č —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā—č —Ä–į–Ĺ–ī–ĺ–ľ–ł–∑–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–≥–ĺ –ī–≤–ĺ–Ļ–Ĺ–ĺ–≥–ĺ —Ā–Ľ–Ķ–Ņ–ĺ–≥–ĺ –Ņ–Ľ–į—Ü–Ķ–Ī–ĺ–ļ–ĺ–Ĺ—ā—Ä–ĺ–Ľ–ł—Ä—É–Ķ–ľ–ĺ–≥–ĺ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź III —Ą–į–∑—č NCIC CTG —Ā¬†—É—á–į—Ā—ā–ł–Ķ–ľ 569 –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤, –ī–ĺ–ļ–į–∑–į–≤—ą–Ķ–≥–ĺ —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć —ć—Ä–Ľ–ĺ—ā–ł–Ĺ–ł–Ī–į –Ņ—Ä–ł –†–ü–Ė. –ü–į—Ü–ł–Ķ–Ĺ—ā—č –Ņ–ĺ–Ľ—É—á–į–Ľ–ł –ļ–ĺ–ľ–Ī–ł–Ĺ–į—Ü–ł—é —ć—Ä–Ľ–ĺ—ā–ł–Ĺ–ł–Ī–į (–Ņ–ĺ 100‚Äď150 –ľ–≥/—Ā—É—ā) —Ā¬†–≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ–ĺ–ľ (7 –Ķ–∂–Ķ–Ĺ–Ķ–ī–Ķ–Ľ—Ć–Ĺ—č—Ö –ī–ĺ–∑ –Ņ–ĺ 1 –≥/–ľ2) –ł–Ľ–ł –ľ–ĺ–Ĺ–ĺ—ā–Ķ—Ä–į–Ņ–ł—é –≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ–ĺ–ľ. –ú–Ķ–ī–ł–į–Ĺ–į –ĺ–Ī—Č–Ķ–Ļ –Ņ—Ä–ĺ–ī–ĺ–Ľ–∂–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ—Ā—ā–ł –∂–ł–∑–Ĺ–ł –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö, –Ņ–ĺ–Ľ—É—á–į–≤—ą–ł—Ö –ĺ–Ī–į –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į, –ĺ–ļ–į–∑–į–Ľ–į—Ā—Ć –≤—č—ą–Ķ: 6,24 –ł¬†5,9 –ľ–Ķ—Ā—Ź—Ü–į —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ. –†–į–∑–Ĺ–ł—Ü–į —Ā–ĺ—Ā—ā–į–≤–ł–Ľ–į –≤—Ā–Ķ–≥–ĺ –Ľ–ł—ą—Ć
0,33 –ľ–Ķ—Ā—Ź—Ü–į, –ĺ–ī–Ĺ–į–ļ–ĺ –ĺ–ļ–į–∑–į–Ľ–į—Ā—Ć —Ā—ā–į—ā–ł—Ā—ā–ł—á–Ķ—Ā–ļ–ł –ī–ĺ—Ā—ā–ĺ–≤–Ķ—Ä–Ĺ–ĺ–Ļ (p = 0,038), —á—ā–ĺ –Ņ–ĺ–∑–≤–ĺ–Ľ–ł–Ľ–ĺ FDA —Ä–į–∑—Ä–Ķ—ą–ł—ā—Ć –Ņ—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł–Ķ —ć—Ä–Ľ–ĺ—ā–ł–Ĺ–ł–Ī–į –Ņ—Ä–ł –ľ–Ķ—ā–į—Ā—ā–į—ā–ł—á–Ķ—Ā–ļ–ĺ–ľ –†–ü–Ė.
–ě—ā–ľ–Ķ—ā–ł–ľ, —á—ā–ĺ –≤¬†—Ö–ĺ–ī–Ķ –ī–į–Ĺ–Ĺ–ĺ–≥–ĺ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź –Ĺ–Ķ –Ī—č–Ľ–ĺ –≤—č—Ź–≤–Ľ–Ķ–Ĺ–ĺ –ļ–ĺ—Ä—Ä–Ķ–Ľ—Ź—Ü–ł–ł –ľ–Ķ–∂–ī—É —É—Ä–ĺ–≤–Ĺ–Ķ–ľ —ć–ļ—Ā–Ņ—Ä–Ķ—Ā—Ā–ł–ł EGFR –ł¬†–ĺ—ā–≤–Ķ—ā–ĺ–ľ –Ĺ–į —ā–Ķ—Ä–į–Ņ–ł—é —ć—Ä–Ľ–ĺ—ā–ł–Ĺ–ł–Ī–ĺ–ľ. –†–į–∑–≤–ł—ā–ł–Ķ —ɬ†–Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā—č–Ņ–ł 2-–Ļ —Ā—ā–Ķ–Ņ–Ķ–Ĺ–ł –≤—č—Ä–į–∂–Ķ–Ĺ–Ĺ–ĺ—Ā—ā–ł –ł¬†–≤—č—ą–Ķ —Ź–≤–Ľ—Ź–Ľ–ĺ—Ā—Ć –Ī–Ľ–į–≥–ĺ–Ņ—Ä–ł—Ź—ā–Ĺ—č–ľ –Ņ—Ä–ĺ–≥–Ĺ–ĺ—Ā—ā–ł—á–Ķ—Ā–ļ–ł–ľ —Ą–į–ļ—ā–ĺ—Ä–ĺ–ľ. –ú–Ķ–ī–ł–į–Ĺ–į –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā–ł –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā¬†—Ā—č–Ņ—Ć—é 0, 1 –ł¬†‚Č• 2-–Ļ —Ā—ā–Ķ–Ņ–Ķ–Ĺ–ł —Ā–ĺ—Ā—ā–į–≤–ł–Ľ–į 5,3, 5,8 –ł¬†10,5 –ľ–Ķ—Ā—Ź—Ü–į —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ, –į¬†–ĺ–ī–Ĺ–ĺ–Ľ–Ķ—ā–Ĺ—Ź—Ź –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā—Ƭ†‚Äď 16, 9 –ł¬†43% [45].
–ö–ĺ–ľ–Ī–ł–Ĺ–į—Ü–ł—é —ć—Ä–Ľ–ĺ—ā–ł–Ĺ–ł–Ī–į –ł¬†–ļ–į–Ņ–Ķ—Ü–ł—ā–į–Ī–ł–Ĺ–į –ł–∑—É—á–į–Ľ–ł –≤¬†–ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–ł II —Ą–į–∑—č (30 –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤) –≤¬†–ļ–į—á–Ķ—Ā—ā–≤–Ķ —Ä–Ķ–∂–ł–ľ–į –≤—ā–ĺ—Ä–ĺ–Ļ –Ľ–ł–Ĺ–ł–ł —ā–Ķ—Ä–į–Ņ–ł–ł –†–ü–Ė, —Ä–Ķ–∑–ł—Ā—ā–Ķ–Ĺ—ā–Ĺ–ĺ–ľ –ļ¬†–≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ—É. –ě–Ī—ä–Ķ–ļ—ā–ł–≤–Ĺ—č–Ļ –ĺ—ā–≤–Ķ—ā –Ī—č–Ľ –∑–į—Ä–Ķ–≥–ł—Ā—ā—Ä–ł—Ä–ĺ–≤–į–Ĺ —ɬ†10% –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤, –ľ–Ķ–ī–ł–į–Ĺ–į –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā–ł —Ā–ĺ—Ā—ā–į–≤–ł–Ľ–į 6,5 –ľ–Ķ—Ā—Ź—Ü–į [46].
–í —Ä–į–Ĺ–ī–ĺ–ľ–ł–∑–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–ľ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–ł III —Ą–į–∑—č S0205, –Ņ—Ä–ĺ–≤–Ķ–ī–Ķ–Ĺ–Ĺ–ĺ–ľ –ģ–≥–ĺ-–∑–į–Ņ–į–ī–Ĺ–ĺ–Ļ –≥—Ä—É–Ņ–Ņ–ĺ–Ļ –ĺ–Ĺ–ļ–ĺ–Ľ–ĺ–≥–ĺ–≤ (Southwest Oncology Group, SWOG), 745 –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā¬†–ľ–Ķ—Ā—ā–Ĺ–ĺ—Ä–į—Ā–Ņ—Ä–ĺ—Ā—ā—Ä–į–Ĺ–Ķ–Ĺ–Ĺ–ĺ–Ļ –Ĺ–Ķ—Ä–Ķ–∑–Ķ–ļ—ā–į–Ī–Ķ–Ľ—Ć–Ĺ–ĺ–Ļ –ĺ–Ņ—É—Ö–ĺ–Ľ—Ć—é –ł–Ľ–ł –ľ–Ķ—ā–į—Ā—ā–į—ā–ł—á–Ķ—Ā–ļ–ł–ľ –†–ü–Ė –Ņ–ĺ–Ľ—É—á–į–Ľ–ł –≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ –≤¬†–ľ–ĺ–Ĺ–ĺ—ā–Ķ—Ä–į–Ņ–ł–ł –ł–Ľ–ł –≤¬†—Ā–ĺ—á–Ķ—ā–į–Ĺ–ł–ł —Ā¬†—Ü–Ķ—ā—É–ļ—Ā–ł–ľ–į–Ī–ĺ–ľ. –°—ā–į—ā–ł—Ā—ā–ł—á–Ķ—Ā–ļ–ł –∑–Ĺ–į—á–ł–ľ–ĺ–Ļ —Ä–į–∑–Ĺ–ł—Ü—č –≤¬†–ľ–Ķ–ī–ł–į–Ĺ–Ķ –ĺ–Ī—Č–Ķ–Ļ –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā–ł (6,9 –ł¬†5,9 –ľ–Ķ—Ā—Ź—Ü–į —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ, —Ä = 0,14), –ľ–Ķ–ī–ł–į–Ĺ–Ķ –≤—Ä–Ķ–ľ–Ķ–Ĺ–ł –ī–ĺ –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź (3,0 –ł¬†3,5 –ľ–Ķ—Ā—Ź—Ü–į, —Ä = 0,058) –ł¬†—á–į—Ā—ā–ĺ—ā–Ķ –ĺ–Ī—ä–Ķ–ļ—ā–ł–≤–Ĺ—č—Ö –ĺ—ā–≤–Ķ—ā–ĺ–≤ –ľ–Ķ–∂–ī—É –ī–≤—É–ľ—Ź –≥—Ä—É–Ņ–Ņ–į–ľ–ł –Ĺ–Ķ –≤—č—Ź–≤–Ľ–Ķ–Ĺ–ĺ. –ü—Ä–ł —ć—ā–ĺ–ľ –≤¬†90% —Ā–Ľ—É—á–į–Ķ–≤ –Ī—č–Ľ–į –ĺ–Ī–Ĺ–į—Ä—É–∂–Ķ–Ĺ–į —ć–ļ—Ā–Ņ—Ä–Ķ—Ā—Ā–ł—Ź EGFR –≤¬†–ĺ–Ņ—É—Ö–ĺ–Ľ–ł, —á—ā–ĺ –Ĺ–ł–ļ–į–ļ –Ĺ–Ķ –Ņ–ĺ–≤–Ľ–ł—Ź–Ľ–ĺ –Ĺ–į —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź [47].
–°—É—Č–Ķ—Ā—ā–≤—É–Ķ—ā –Ĺ–Ķ—Ā–ļ–ĺ–Ľ—Ć–ļ–ĺ –≥–ł–Ņ–ĺ—ā–Ķ–∑, –ĺ–Ī—ä—Ź—Ā–Ĺ—Ź—é—Č–ł—Ö —Ä–Ķ–∑–ł—Ā—ā–Ķ–Ĺ—ā–Ĺ–ĺ—Ā—ā—Ć –į–ī–Ķ–Ĺ–ĺ–ļ–į—Ä—Ü–ł–Ĺ–ĺ–ľ—č –Ņ–ĺ–ī–∂–Ķ–Ľ—É–ī–ĺ—á–Ĺ–ĺ–Ļ –∂–Ķ–Ľ–Ķ–∑—č –ļ¬†–į–Ĺ—ā–ł-EGFR-—ā–Ķ—Ä–į–Ņ–ł–ł. –Ě–į –Ņ—Ä–ł–ľ–Ķ—Ä–Ķ –Ĺ–Ķ–ľ–Ķ–Ľ–ļ–ĺ–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ–ĺ–≥–ĺ —Ä–į–ļ–į –Ľ–Ķ–≥–ļ–ĺ–≥–ĺ –Ī—č–Ľ–ĺ –Ņ–ĺ–ļ–į–∑–į–Ĺ–ĺ, —á—ā–ĺ –Ĺ–į–Ľ–ł—á–ł–Ķ –ľ—É—ā–į—Ü–ł–ł –≤¬†—ā–ł—Ä–ĺ–∑–ł–Ĺ–ļ–ł–Ĺ–į–∑–Ĺ–ĺ–ľ –ī–ĺ–ľ–Ķ–Ĺ–Ķ EGFR¬†‚Äď –Ņ—Ä–ĺ–≥–Ĺ–ĺ—Ā—ā–ł—á–Ķ—Ā–ļ–ł–Ļ —Ą–į–ļ—ā–ĺ—Ä –≤¬†–ĺ—ā–Ĺ–ĺ—ą–Ķ–Ĺ–ł–ł –ĺ—ā–≤–Ķ—ā–į –Ĺ–į –Ľ–Ķ—á–Ķ–Ĺ–ł–Ķ —ć—Ä–Ľ–ĺ—ā–ł–Ĺ–ł–Ī–ĺ–ľ. –°—É–ī—Ź –Ņ–ĺ –Ņ–ĺ—Ā–Ľ–Ķ–ī–Ĺ–ł–ľ –ī–į–Ĺ–Ĺ—č–ľ, –ľ—É—ā–į—Ü–ł—Ź –≤¬†–≥–Ķ–Ĺ–Ķ EGFR –ļ—Ä–į–Ļ–Ĺ–Ķ —Ä–Ķ–ī–ļ–ĺ –≤—Ā—ā—Ä–Ķ—á–į–Ķ—ā—Ā—Ź –Ņ—Ä–ł –†–ü–Ė [48, 49].
–ü—Ä–ł –Ī–Ľ–ĺ–ļ–ł—Ä–ĺ–≤–į–Ĺ–ł–ł —ā–ł—Ä–ĺ–∑–ł–Ĺ–ļ–ł–Ĺ–į–∑—č EGFR –ĺ—Ā—ā–į–Ķ—ā—Ā—Ź –≤–ĺ–∑–ľ–ĺ–∂–Ĺ—č–ľ –į–ļ—ā–ł–≤–ł–∑–ł—Ä–ĺ–≤–į—ā—Ć 2 –≥–Ľ–į–≤–Ĺ—č—Ö –≤–Ĺ—É—ā—Ä–ł–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—č—Ö —Ā–ł–≥–Ĺ–į–Ľ—Ć–Ĺ—č—Ö –Ņ—É—ā–ł (PI3K/Akt –ł¬†MAPK) —á–Ķ—Ä–Ķ–∑ —ā–ł—Ä–ĺ–∑–ł–Ĺ–ļ–ł–Ĺ–į–∑—č —Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–ĺ–≤ –ļ¬†–ī—Ä—É–≥–ł–ľ —Ä–ĺ—Ā—ā–ĺ–≤—č–ľ —Ą–į–ļ—ā–ĺ—Ä–į–ľ.
–ö–į–ļ —É–Ņ–ĺ–ľ–ł–Ĺ–į–Ľ–ĺ—Ā—Ć –≤—č—ą–Ķ, –į–ļ—ā–ł–≤–ł—Ä—É—é—Č–į—Ź –ľ—É—ā–į—Ü–ł—Ź –≥–Ķ–Ĺ–į KRAS –ĺ–Ņ—Ä–Ķ–ī–Ķ–Ľ—Ź–Ķ—ā—Ā—Ź —É–∂–Ķ –Ĺ–į —Ä–į–Ĺ–Ĺ–ł—Ö —Ā—ā–į–ī–ł—Ź—Ö –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź –≤¬†85% —Ā–Ľ—É—á–į–Ķ–≤ [50]. –ü—Ä–ł –Ĺ–į–Ľ–ł—á–ł–ł –ľ—É—ā–į—Ü–ł–ł –≤¬†–≥–Ķ–Ĺ–Ķ KRAS –į–Ĺ—ā–ł-EGFR-—ā–Ķ—Ä–į–Ņ–ł—Ź —Ā—ā–į–Ĺ–ĺ–≤–ł—ā—Ā—Ź –Ĺ–Ķ—ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ–Ļ [51, 52].
–¶–Ķ—ā—É–ļ—Ā–ł–ľ–į–Ī –ĺ–ļ–į–∑—č–≤–į–Ķ—ā —Ü–ł—ā–ĺ—ā–ĺ–ļ—Ā–ł—á–Ķ—Ā–ļ–ĺ–Ķ –ī–Ķ–Ļ—Ā—ā–≤–ł–Ķ –Ĺ–į –∑–Ľ–ĺ–ļ–į—á–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ—É—é –ļ–Ľ–Ķ—ā–ļ—É —ā–į–ļ–∂–Ķ –∑–į —Ā—á–Ķ—ā –ł–ľ–ľ—É–Ĺ–ĺ–ĺ–Ņ–ĺ—Ā—Ä–Ķ–ī–ĺ–≤–į–Ĺ–Ĺ–ĺ–≥–ĺ –Ņ—Ä–ĺ—ā–ł–≤–ĺ–ĺ–Ņ—É—Ö–ĺ–Ľ–Ķ–≤–ĺ–≥–ĺ —ć—Ą—Ą–Ķ–ļ—ā–į, —Ä–Ķ–į–Ľ–ł–∑—É—é—Č–Ķ–≥–ĺ—Ā—Ź –∑–į —Ā—á–Ķ—ā —Ā–≤—Ź–∑—č–≤–į–Ĺ–ł—Ź Fc-—Ą—Ä–į–≥–ľ–Ķ–Ĺ—ā–į —Ö–ł–ľ–Ķ—Ä–Ĺ–ĺ–≥–ĺ –ľ–ĺ–Ĺ–ĺ–ļ–Ľ–ĺ–Ĺ–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –į–Ĺ—ā–ł—ā–Ķ–Ľ–į —Ā¬†Fc-—Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–į–ľ–ł (Fc-–≥–į–ľ–ľ–į-Rs) –Ĺ–į –Ņ–ĺ–≤–Ķ—Ä—Ö–Ĺ–ĺ—Ā—ā–ł –ł–ľ–ľ—É–Ĺ–Ĺ—č—Ö —ć—Ą—Ą–Ķ–ļ—ā–ĺ—Ä–Ĺ—č—Ö –ļ–Ľ–Ķ—ā–ĺ–ļ. –ü—Ä–ł –Ņ–ĺ–Ľ–ł–ľ–ĺ—Ä—Ą–ł–∑–ľ–Ķ —Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–ĺ–≤ (Fc-–≥–į–ľ–ľ–į-RIIa –ł¬†Fc-–≥–į–ľ–ľ–į-RIIIa) –Ľ–ł–ľ—Ą–ĺ—Ü–ł—ā–ĺ–≤ —É–ľ–Ķ–Ĺ—Ć—ą–į–Ķ—ā—Ā—Ź –ł—Ö –į—Ą—Ą–ł–Ĺ–Ĺ–ĺ—Ā—ā—Ć –ļ¬†Ig G1 (—Ü–Ķ—ā—É–ļ—Ā–ł–ľ–į–Ī), —á—ā–ĺ –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ¬†—Ā–Ĺ–ł–∂–Ķ–Ĺ–ł—é —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź [53].
–°—É—Č–Ķ—Ā—ā–≤—É–Ķ—ā –≥–ł–Ņ–ĺ—ā–Ķ–∑–į, —Ā–ĺ–≥–Ľ–į—Ā–Ĺ–ĺ –ļ–ĺ—ā–ĺ—Ä–ĺ–Ļ –ĺ–Ņ—É—Ö–ĺ–Ľ–Ķ–≤—č–Ķ –ļ–Ľ–Ķ—ā–ļ–ł –ľ–ĺ–≥—É—ā –Ņ–Ķ—Ä–Ķ–ļ–Ľ—é—á–į—ā—Ć —Ä–į–∑–Ľ–ł—á–Ĺ—č–Ķ –ľ–ĺ–Ľ–Ķ–ļ—É–Ľ—Ź—Ä–Ĺ—č–Ķ –Ņ—É—ā–ł –Ņ–Ķ—Ä–Ķ–ī–į—á–ł –ľ–ł—ā–ĺ–≥–Ķ–Ĺ–Ĺ–ĺ–≥–ĺ —Ā–ł–≥–Ĺ–į–Ľ–į. –í¬†—Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–ł–ł —Ā¬†—ć—ā–ĺ–Ļ –ļ–ĺ–Ĺ—Ü–Ķ–Ņ—Ü–ł–Ķ–Ļ —ć—Ä–Ľ–ĺ—ā–ł–Ĺ–ł–Ī –Ņ–ĺ–Ľ–Ĺ–ĺ—Ā—ā—Ć—é –Ī–Ľ–ĺ–ļ–ł—Ä—É–Ķ—ā EGFR-–ĺ–Ņ–ĺ—Ā—Ä–Ķ–ī–ĺ–≤–į–Ĺ–Ĺ—č–Ļ –Ņ—É—ā—Ć, –Ĺ–ĺ –Ņ—Ä–ł —ć—ā–ĺ–ľ –Ĺ–Ķ –ĺ–ļ–į–∑—č–≤–į–Ķ—ā –≤–Ľ–ł—Ź–Ĺ–ł—Ź –Ĺ–į –Ņ—Ä–ĺ–Ľ–ł—Ą–Ķ—Ä–į—Ü–ł—é –ļ–Ľ–Ķ—ā–ļ–ł, –į–ļ—ā–ł–≤–ł—Ä–ĺ–≤–į–Ĺ–Ĺ—É—é –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–ĺ–Ņ–ĺ–ī–ĺ–Ī–Ĺ—č–ľ —Ą–į–ļ—ā–ĺ—Ä–ĺ–ľ —Ä–ĺ—Ā—ā–į [54].
–°–ĺ—Ā—É–ī–ł—Ā—ā—č–Ļ —ć–Ĺ–ī–ĺ—ā–Ķ–Ľ–ł–į–Ľ—Ć–Ĺ—č–Ļ —Ą–į–ļ—ā–ĺ—Ä —Ä–ĺ—Ā—ā–į
–í–į–∂–Ĺ—č–ľ —É—Ā–Ľ–ĺ–≤–ł–Ķ–ľ —Ą–ĺ—Ä–ľ–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź –Ņ–Ķ—Ä–≤–ł—á–Ĺ–ĺ–Ļ –ĺ–Ņ—É—Ö–ĺ–Ľ–ł –ł¬†–ľ–Ķ—ā–į—Ā—ā–į–∑–ĺ–≤ —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –Ĺ–Ķ–ĺ–į–Ĺ–≥–ł–ĺ–≥–Ķ–Ĺ–Ķ–∑¬†‚Äď —Ą–ĺ—Ä–ľ–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ –ī–ĺ–Ņ–ĺ–Ľ–Ĺ–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ļ –ļ–į–Ņ–ł–Ľ–Ľ—Ź—Ä–Ĺ–ĺ–Ļ —Ā–Ķ—ā–ł, –ĺ–Ī–Ķ—Ā–Ņ–Ķ—á–ł–≤–į—é—Č–Ķ–Ļ —Ä–ĺ—Ā—ā –ĺ–Ņ—É—Ö–ĺ–Ľ–Ķ–≤–ĺ–≥–ĺ —É–∑–Ľ–į. –Ď–Ķ–Ľ–ļ–ł —Ā–Ķ–ľ–Ķ–Ļ—Ā—ā–≤–į —Ā–ĺ—Ā—É–ī–ł—Ā—ā–ĺ–≥–ĺ —ć–Ĺ–ī–ĺ—ā–Ķ–Ľ–ł–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ —Ą–į–ļ—ā–ĺ—Ä–į —Ä–ĺ—Ā—ā–į (VEGF) –ł¬†—Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä—č –ļ¬†–Ĺ–ł–ľ –ĺ–Ī–Ķ—Ā–Ņ–Ķ—á–ł–≤–į—é—ā –ĺ–Ī—Ä–į–∑–ĺ–≤–į–Ĺ–ł–Ķ –Ĺ–ĺ–≤—č—Ö —Ā–ĺ—Ā—É–ī–ĺ–≤, —á—ā–ĺ –Ĺ–Ķ–ĺ–Ī—Ö–ĺ–ī–ł–ľ–ĺ –ī–Ľ—Ź —Ä–ĺ—Ā—ā–į –ĺ–Ņ—É—Ö–ĺ–Ľ–ł. –ď–ł–Ņ–Ķ—Ä—ć–ļ—Ā–Ņ—Ä–Ķ—Ā—Ā–ł—Ź VEGF –Ĺ–į–Ī–Ľ—é–ī–į–Ķ—ā—Ā—Ź –≤¬†64% –ļ–Ľ–Ķ—ā–ĺ–ļ –†–ü–Ė [55]. –ü—Ä–ĺ–ī–ĺ–Ľ–∂–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ—Ā—ā—Ć –∂–ł–∑–Ĺ–ł –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤, –≤¬†–ĺ–Ņ—É—Ö–ĺ–Ľ–ł –ļ–ĺ—ā–ĺ—Ä—č—Ö –ĺ–Ņ—Ä–Ķ–ī–Ķ–Ľ—Ź–Ķ—ā—Ā—Ź –≤—č—Ā–ĺ–ļ–ł–Ļ —É—Ä–ĺ–≤–Ķ–Ĺ—Ć —ć–ļ—Ā–Ņ—Ä–Ķ—Ā—Ā–ł–ł VEGF, –ī–ĺ—Ā—ā–ĺ–≤–Ķ—Ä–Ĺ–ĺ –ľ–Ķ–Ĺ—Ć—ą–Ķ, —á–Ķ–ľ —ɬ†–Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ –Ī–Ķ–∑ –≤—č—Ā–ĺ–ļ–ĺ–≥–ĺ —É—Ä–ĺ–≤–Ĺ—Ź —ć–ļ—Ā–Ņ—Ä–Ķ—Ā—Ā–ł–ł VEGF [56]. –Ě–Ķ—Ā–ľ–ĺ—ā—Ä—Ź –Ĺ–į –ľ–Ĺ–ĺ–≥–ĺ–ĺ–Ī–Ķ—Č–į—é—Č–ł–Ķ —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā—č –ī–ĺ–ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö –ł—Ā–Ņ—č—ā–į–Ĺ–ł–Ļ –Ī–Ķ–≤–į—Ü–ł–∑—É–ľ–į–Ī–į, –≤¬†—Ä–į–Ĺ–ī–ĺ–ľ–ł–∑–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–ľ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–ł III —Ą–į–∑—č CALGB 80303 –ľ–Ķ–ī–ł–į–Ĺ—č –ĺ–Ī—Č–Ķ–Ļ –Ņ—Ä–ĺ–ī–ĺ–Ľ–∂–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ—Ā—ā–ł –∂–ł–∑–Ĺ–ł –≤¬†–≥—Ä—É–Ņ–Ņ–į—Ö –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤, –Ņ–ĺ–Ľ—É—á–į–≤—ą–ł—Ö –≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ –≤¬†—Ā–ĺ—á–Ķ—ā–į–Ĺ–ł–ł —Ā¬†–Ņ–Ľ–į—Ü–Ķ–Ī–ĺ –ł–Ľ–ł —Ā¬†–Ī–Ķ–≤–į—Ü–ł–∑—É–ľ–į–Ī–ĺ–ľ, –Ņ—Ä–į–ļ—ā–ł—á–Ķ—Ā–ļ–ł –Ĺ–Ķ —Ä–į–∑–Ľ–ł—á–į–Ľ–ł—Ā—Ć: 5,8 –ł¬†5,9 –ľ–Ķ—Ā—Ź—Ü–į (p = 0,95) [57]. –ú–Ķ–ī–ł–į–Ĺ–į –≤—Ä–Ķ–ľ–Ķ–Ĺ–ł –ī–ĺ –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź —Ā–ĺ—Ā—ā–į–≤–ł–Ľ–į 3,8 –ł¬†2,9 –ľ–Ķ—Ā—Ź—Ü–į —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ (p = 0,07), —á–į—Ā—ā–ĺ—ā–į –ĺ–Ī—ä–Ķ–ļ—ā–ł–≤–Ĺ—č—Ö –ĺ—ā–≤–Ķ—ā–ĺ–≤¬†‚Äď 13 –ł¬†10%.
–í –ī–≤–ĺ–Ļ–Ĺ–ĺ–ľ —Ā–Ľ–Ķ–Ņ–ĺ–ľ —Ä–į–Ĺ–ī–ĺ–ľ–ł–∑–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–ľ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–ł III —Ą–į–∑—č AVITA, –≥–ī–Ķ –ĺ—Ü–Ķ–Ĺ–ł–≤–į–Ľ—Ā—Ź —Ä–Ķ–∂–ł–ľ —ā–Ķ—Ä–į–Ņ–ł–ł –ļ–ĺ–ľ–Ī–ł–Ĺ–į—Ü–ł–Ķ–Ļ –≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ–į –ł¬†—ć—Ä–Ľ–ĺ—ā–ł–Ĺ–ł–Ī–į –≤¬†—Ā–ĺ—á–Ķ—ā–į–Ĺ–ł–ł —Ā¬†–Ī–Ķ–≤–į—Ü–ł–∑—É–ľ–į–Ī–ĺ–ľ –ł¬†–Ī–Ķ–∑ –Ĺ–Ķ–≥–ĺ, –Ī—č–Ľ–ĺ –≤—č—Ź–≤–Ľ–Ķ–Ĺ–ĺ —É–≤–Ķ–Ľ–ł—á–Ķ–Ĺ–ł–Ķ –≤—Ä–Ķ–ľ–Ķ–Ĺ–ł –ī–ĺ –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź (4,6 –ł¬†3,6 –ľ–Ķ—Ā—Ź—Ü–į, p = 0,0002), –Ĺ–ĺ –Ĺ–Ķ –ĺ–Ī—Č–Ķ–Ļ –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā–ł (7,1 –ł¬†6 –ľ–Ķ—Ā—Ź—Ü–Ķ–≤, p = 0,2087) [58].
–ė–Ĺ–≥–ł–Ī–ł—ā–ĺ—Ä—č —Ü–ł–ļ–Ľ–ĺ–ĺ–ļ—Ā–ł–≥–Ķ–Ĺ–į–∑—č 2 —ā–ł–Ņ–į
–¶–ł–ļ–Ľ–ĺ–ĺ–ļ—Ā–ł–≥–Ķ–Ĺ–į–∑–į (–¶–ě–ď) —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź —Ą–Ķ—Ä–ľ–Ķ–Ĺ—ā–ĺ–ľ, –Ņ—Ä–Ķ–≤—Ä–į—Č–į—é—Č–ł–ľ –į—Ä–į—Ö–ł–ī–ĺ–Ĺ–ĺ–≤—É—é –ļ–ł—Ā–Ľ–ĺ—ā—É –≤¬†–Ņ—Ä–ĺ—Ā—ā–į–≥–Ľ–į–Ĺ–ī–ł–Ĺ—č, –Ņ—Ä–ĺ—Ā—ā–į—Ü–ł–ļ–Ľ–ł–Ĺ—č –ł¬†—ā—Ä–ĺ–ľ–Ī–ĺ–ļ—Ā–į–Ĺ—č. –¶–ě–ď –Ņ—Ä–Ķ–ī—Ā—ā–į–≤–Ľ–Ķ–Ĺ–į –ī–≤—É–ľ—Ź –ł–∑–ĺ—Ą–ĺ—Ä–ľ–į–ľ–ł: –¶–ě–ď-1, –ļ–ĺ—ā–ĺ—Ä–į—Ź –Ņ–ĺ—Ā—ā–ĺ—Ź–Ĺ–Ĺ–ĺ —ć–ļ—Ā–Ņ—Ä–Ķ—Ā—Ā–ł—Ä—É–Ķ—ā—Ā—Ź –≤¬†–Ĺ–ĺ—Ä–ľ–į–Ľ—Ć–Ĺ—č—Ö —ā–ļ–į–Ĺ—Ź—Ö, –ł¬†–¶–ě–ď-2, —Ā–ł–Ĺ—ā–Ķ–∑ –ļ–ĺ—ā–ĺ—Ä–ĺ–Ļ –≥–Ľ–į–≤–Ĺ—č–ľ –ĺ–Ī—Ä–į–∑–ĺ–ľ –ł–Ĺ–ī—É—Ü–ł—Ä—É–Ķ—ā—Ā—Ź –Ņ—Ä–ĺ–≤–ĺ—Ā–Ņ–į–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ—č–ľ–ł —Ü–ł—ā–ĺ–ļ–ł–Ĺ–į–ľ–ł, —Ą–į–ļ—ā–ĺ—Ä–į–ľ–ł —Ä–ĺ—Ā—ā–į –ł¬†–ĺ–Ĺ–ļ–ĺ–≥–Ķ–Ĺ–į–ľ–ł. –ü–ĺ–≤—č—ą–Ķ–Ĺ–Ĺ–į—Ź —ć–ļ—Ā–Ņ—Ä–Ķ—Ā—Ā–ł—Ź –¶–ě–ď-2 —á–į—Ā—ā–ĺ –Ĺ–į–Ī–Ľ—é–ī–į–Ķ—ā—Ā—Ź –ł¬†—Ā–Ņ–ĺ—Ā–ĺ–Ī—Ā—ā–≤—É–Ķ—ā –ļ–į–Ĺ—Ü–Ķ—Ä–ĺ–≥–Ķ–Ĺ–Ķ–∑—É –Ņ—Ä–ł –ľ–Ĺ–ĺ–≥–ł—Ö —Ā–ĺ–Ľ–ł–ī–Ĺ—č—Ö –ĺ–Ņ—É—Ö–ĺ–Ľ—Ź—Ö, –≤¬†—ā–ĺ–ľ —á–ł—Ā–Ľ–Ķ –ł¬†–†–ü–Ė, –Ņ–ĺ—ć—ā–ĺ–ľ—É —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –Ņ–Ľ–ĺ—Ö–ł–ľ –Ņ—Ä–ĺ–≥–Ĺ–ĺ—Ā—ā–ł—á–Ķ—Ā–ļ–ł–ľ —Ą–į–ļ—ā–ĺ—Ä–ĺ–ľ [59, 60, 61].
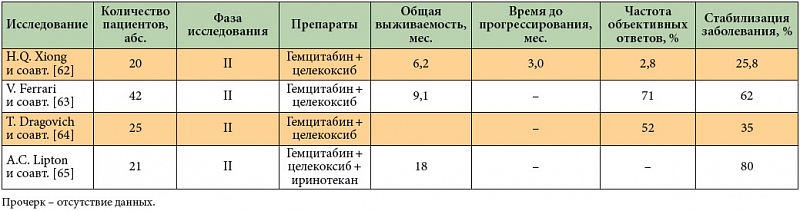
–ė–Ĺ–≥–ł–Ī–ł—ā–ĺ—Ä –¶–ě–ď-2 —Ü–Ķ–Ľ–Ķ–ļ–ĺ–ļ—Ā–ł–Ī –≤¬†–ī–ĺ–ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł—Ö –ł—Ā–Ņ—č—ā–į–Ĺ–ł—Ź—Ö –Ņ—Ä–ĺ—Ź–≤–ł–Ľ —Ā–ł–Ĺ–Ķ—Ä–≥–Ķ—ā–ł—á–Ķ—Ā–ļ–ł–Ļ —ć—Ą—Ą–Ķ–ļ—ā –Ņ—Ä–ł –ļ–ĺ–ľ–Ī–ł–Ĺ–į—Ü–ł–ł —Ā¬†—Ü–ł—ā–ĺ—ā–ĺ–ļ—Ā–ł—á–Ķ—Ā–ļ–ł–ľ–ł –į–≥–Ķ–Ĺ—ā–į–ľ–ł (5-—Ą—ā–ĺ—Ä—É—Ä–į—Ü–ł–Ľ, –≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ), –Ľ—É—á–Ķ–≤–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–Ķ–Ļ –ł¬†—Ā –ī—Ä—É–≥–ł–ľ–ł —ā–į—Ä–≥–Ķ—ā–Ĺ—č–ľ–ł –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į–ľ–ł (—ć—Ä–Ľ–ĺ—ā–ł–Ĺ–ł–Ī, –ļ—É—Ä–ļ—É–ľ–ł–Ĺ). –ě–ī–Ĺ–į–ļ–ĺ —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā—č –Ĺ–Ķ—Ā–ļ–ĺ–Ľ—Ć–ļ–ł—Ö –Ĺ–Ķ–Ī–ĺ–Ľ—Ć—ą–ł—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ II —Ą–į–∑—č –Ņ—Ä–ĺ—ā–ł–≤–ĺ—Ä–Ķ—á–ł–≤—č [62‚Äď65], —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ, —ā—Ä–Ķ–Ī—É—é—ā—Ā—Ź –ī–ĺ–Ņ–ĺ–Ľ–Ĺ–ł—ā–Ķ–Ľ—Ć–Ĺ—č–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź (—ā–į–Ī–Ľ. 4).
–ė–Ĺ—Ā—É–Ľ–ł–Ĺ–ĺ–Ņ–ĺ–ī–ĺ–Ī–Ĺ—č–Ļ —Ą–į–ļ—ā–ĺ—Ä —Ä–ĺ—Ā—ā–į 1 —ā–ł–Ņ–į
–†–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–ĺ–Ņ–ĺ–ī–ĺ–Ī–Ĺ–ĺ–≥–ĺ —Ą–į–ļ—ā–ĺ—Ä–į —Ä–ĺ—Ā—ā–į 1 —ā–ł–Ņ–į (IGFR1) –Ņ—Ä–Ķ–ī—Ā—ā–į–≤–Ľ—Ź–Ķ—ā —Ā–ĺ–Ī–ĺ–Ļ —ā—Ä–į–Ĺ—Ā–ľ–Ķ–ľ–Ī—Ä–į–Ĺ–Ĺ—č–Ļ —ā–ł—Ä–ĺ–∑–ł–Ĺ–ļ–ł–Ĺ–į–∑–Ĺ—č–Ļ —Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä, –≥–ł–Ņ–Ķ—Ä—ć–ļ—Ā–Ņ—Ä–Ķ—Ā—Ā–ł—Ź –ļ–ĺ—ā–ĺ—Ä–ĺ–≥–ĺ –Ĺ–į–Ī–Ľ—é–ī–į–Ķ—ā—Ā—Ź –≤¬†64% —Ā–Ľ—É—á–į–Ķ–≤ –†–ü–Ė [66]. –í¬†–Ĺ–į—Ā—ā–ĺ—Ź—Č–Ķ–Ķ –≤—Ä–Ķ–ľ—Ź –Ņ—Ä–ĺ–≤–ĺ–ī—Ź—ā—Ā—Ź —Ä–į–Ĺ–ī–ĺ–ľ–ł–∑–ł—Ä–ĺ–≤–į–Ĺ–Ĺ—č–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź II —Ą–į–∑—č, –≤¬†–ļ–ĺ—ā–ĺ—Ä—č—Ö –ł–∑—É—á–į–Ķ—ā—Ā—Ź —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć –ł–Ĺ–≥–ł–Ī–ł—ā–ĺ—Ä–ĺ–≤ IGFR1, –ľ–ĺ–Ľ–Ķ–ļ—É–Ľ AMG 479 –ł¬†MK-0646 [67, 68].
–ú—É—ā–į—Ü–ł—Ź –≥–Ķ–Ĺ–į BRCA 2
–ü–ĺ —Ä–į–∑–Ĺ—č–ľ –ī–į–Ĺ–Ĺ—č–ľ, –≥–Ķ—Ä–ľ–ł–Ĺ–į–Ľ—Ć–Ĺ—č–Ķ –ľ—É—ā–į—Ü–ł–ł –≥–Ķ–Ĺ–į BRCA 2, –≥–Ľ–į–≤–Ĺ—č–ľ –ĺ–Ī—Ä–į–∑–ĺ–ľ 6174delT –ł¬†6158insT, –≤—Ā—ā—Ä–Ķ—á–į—é—ā—Ā—Ź —Ā¬†—á–į—Ā—ā–ĺ—ā–ĺ–Ļ –ĺ—ā 6 –ī–ĺ 17% —Ā–Ľ—É—á–į–Ķ–≤ —Ā–Ķ–ľ–Ķ–Ļ–Ĺ–ĺ–≥–ĺ –†–ü–Ė [69‚Äď72] –ł¬†–≤ 10% —Ā–Ņ–ĺ—Ä–į–ī–ł—á–Ķ—Ā–ļ–ł—Ö —Ā–Ľ—É—á–į–Ķ–≤ [73, 74]. –ě–Ņ—É—Ö–ĺ–Ľ–Ķ–≤—č–Ķ –ļ–Ľ–Ķ—ā–ļ–ł —Ā¬†–ľ—É—ā–į—Ü–ł—Ź–ľ–ł –≤¬†–≥–Ķ–Ĺ–į—Ö BRCA 1 / BRCA 2 –≤—č—Ā–ĺ–ļ–ĺ—á—É–≤—Ā—ā–≤–ł—ā–Ķ–Ľ—Ć–Ĺ—č –ļ¬†–≤–ĺ–∑–ī–Ķ–Ļ—Ā—ā–≤–ł—é –į–≥–Ķ–Ĺ—ā–ĺ–≤-–ļ—Ä–ĺ—Ā—Ā–Ľ–ł–Ĺ–ļ–Ķ—Ä–ĺ–≤ (–ú–ł—ā–ĺ–ľ–ł—Ü–ł–Ĺ –°,
–Ņ—Ä–Ķ–Ņ–į—Ä–į—ā—č –Ņ–Ľ–į—ā–ł–Ĺ—č). –ě–Ņ–ł—Ā–į–Ĺ—č —Ā–Ľ—É—á–į–ł, –ļ–ĺ–≥–ī–į —ɬ†–Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤¬†‚Äď –Ĺ–ĺ—Ā–ł—ā–Ķ–Ľ–Ķ–Ļ –ľ—É—ā–į—Ü–ł–Ļ BRCA –Ĺ–į —Ą–ĺ–Ĺ–Ķ –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į–ľ–ł –Ņ–Ľ–į—ā–ł–Ĺ—č –Ĺ–į—Ā—ā—É–Ņ–į–Ľ–į –Ņ–ĺ–Ľ–Ĺ–į—Ź —Ä–Ķ–≥—Ä–Ķ—Ā—Ā–ł—Ź –ĺ–Ņ—É—Ö–ĺ–Ľ–ł –Ņ–ĺ–ī–∂–Ķ–Ľ—É–ī–ĺ—á–Ĺ–ĺ–Ļ –∂–Ķ–Ľ–Ķ–∑—č [75]. –í¬†—Ā–≤—Ź–∑–ł —Ā¬†—ć—ā–ł–ľ –ĺ–ī–Ĺ–ł–ľ –ł–∑ –Ņ–Ķ—Ä—Ā–Ņ–Ķ–ļ—ā–ł–≤–Ĺ—č—Ö –Ņ—É—ā–Ķ–Ļ —Ä–į–∑–≤–ł—ā–ł—Ź —ā–į—Ä–≥–Ķ—ā–Ĺ–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł –†–ü–Ė —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –ł–∑—É—á–Ķ–Ĺ–ł–Ķ PARP-–ł–Ĺ–≥–ł–Ī–ł—ā–ĺ—Ä–ĺ–≤ (–ĺ–Ľ–į–Ņ–į—Ä–ł–Ī, –≤–Ķ–Ľ–ł–Ņ–į—Ä–ł–Ī, BSI-201).
–ó–į–ļ–Ľ—é—á–Ķ–Ĺ–ł–Ķ
–Ě–į —Ā–Ķ–≥–ĺ–ī–Ĺ—Ź—ą–Ĺ–ł–Ļ –ī–Ķ–Ĺ—Ć –Ĺ–ł –ĺ–ī–ł–Ĺ –ł–∑ —Ö–ł–ľ–ł–ĺ—ā–Ķ—Ä–į–Ņ–Ķ–≤—ā–ł—á–Ķ—Ā–ļ–ł—Ö —Ä–Ķ–∂–ł–ľ–ĺ–≤ –Ĺ–Ķ —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź —ć—ā–į–Ľ–ĺ–Ĺ–ĺ–ľ –≤¬†–Ľ–Ķ—á–Ķ–Ĺ–ł–ł –ī–ł—Ā—Ā–Ķ–ľ–ł–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–≥–ĺ –†–ü–Ė.
–ó–į –Ņ–ĺ—Ā–Ľ–Ķ–ī–Ĺ–ł–Ķ –≥–ĺ–ī—č –Ī—č–Ľ–ĺ –ĺ—Ü–Ķ–Ĺ–Ķ–Ĺ–ĺ –Ī–ĺ–Ľ–Ķ–Ķ 33 —Ä–į–Ĺ–ī–ĺ–ľ–ł–∑–ł—Ä–ĺ–≤–į–Ĺ–Ĺ—č—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ —Ā¬†—É—á–į—Ā—ā–ł–Ķ–ľ 6026 –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö. –°—Ä–į–≤–Ĺ–Ķ–Ĺ–ł–Ķ —Ā–ł–ľ–Ņ—ā–ĺ–ľ–į—ā–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź –ł¬†—Ö–ł–ľ–ł–ĺ—ā–Ķ—Ä–į–Ņ–ł–ł –≤—č—Ź–≤–ł–Ľ–ĺ –Ņ—Ä–Ķ–ł–ľ—É—Č–Ķ—Ā—ā–≤–ĺ –Ņ–ĺ—Ā–Ľ–Ķ–ī–Ĺ–Ķ–Ļ —Ā¬†—ā–ĺ—á–ļ–ł –∑—Ä–Ķ–Ĺ–ł—Ź –Ņ—Ä–ĺ–ī–ĺ–Ľ–∂–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ—Ā—ā–ł –∂–ł–∑–Ĺ–ł: —Ä–ł—Ā–ļ —Ā–ľ–Ķ—Ä—ā–ł —Ā–Ĺ–ł–∑–ł–Ľ—Ā—Ź –Ĺ–į 36%. –Ē–ĺ–ļ–į–∑–į–Ĺ–ĺ, —á—ā–ĺ –Ņ—Ä–ĺ–ī–ĺ–Ľ–∂–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ—Ā—ā—Ć –∂–ł–∑–Ĺ–ł —É–≤–Ķ–Ľ–ł—á–ł–≤–į–Ķ—ā—Ā—Ź –Ņ—Ä–ł –Ņ–ĺ–Ľ–ł—Ö–ł–ľ–ł–ĺ—ā–Ķ—Ä–į–Ņ–ł–ł, –≤–ļ–Ľ—é—á–į—é—Č–Ķ–Ļ –≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ, –Ņ–ĺ —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—é —Ā¬†–ľ–ĺ–Ĺ–ĺ—ā–Ķ—Ä–į–Ņ–ł–Ķ–Ļ –≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ–ĺ–ľ: —Ä–ł—Ā–ļ —Ā–ľ–Ķ—Ä—ā–ł —Ā–Ĺ–ł–∑–ł–Ľ—Ā—Ź –Ĺ–į 9% (14 –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ —Ā¬†—É—á–į—Ā—ā–ł–Ķ–ľ 4060 –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö). –Ě–į–ł–Ī–ĺ–Ľ–Ķ–Ķ –į–ļ—ā–ł–≤–Ĺ—č–ľ–ł –ī—É–Ņ–Ľ–Ķ—ā–į–ľ–ł, –≤–Ľ–ł—Ź—é—Č–ł–ľ–ł –Ĺ–į —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć —ā–Ķ—Ä–į–Ņ–ł–ł –ł¬†–Ĺ–į –≤—Ä–Ķ–ľ—Ź –ī–ĺ –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź, —Ź–≤–Ľ—Ź—é—ā—Ā—Ź –ļ–ĺ–ľ–Ī–ł–Ĺ–į—Ü–ł–ł –≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ–į –ł¬†–Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤ –Ņ–Ľ–į—ā–ł–Ĺ—č (3 –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź, 1077 –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö) –ł–Ľ–ł –ļ–į–Ņ–Ķ—Ü–ł—ā–į–Ī–ł–Ĺ–į (3 –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź, 935 –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö). –Ě–į–ł–Ľ—É—á—ą–ł–Ķ —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā—č –Ņ–ĺ –Ņ—Ä–ĺ–ī–ĺ–Ľ–∂–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ—Ā—ā–ł –∂–ł–∑–Ĺ–ł –Ņ–ĺ–Ľ—É—á–Ķ–Ĺ—č —É¬†–Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤, –ĺ–Ī—Č–Ķ–Ķ —Ā–ĺ—Ā—ā–ĺ—Ź–Ĺ–ł–Ķ –ļ–ĺ—ā–ĺ—Ä—č—Ö –ĺ—Ü–Ķ–Ĺ–ł–≤–į–Ľ–ĺ—Ā—Ć –ļ–į–ļ ¬ę—Ö–ĺ—Ä–ĺ—ą–Ķ–Ķ¬Ľ. –ü—Ä–ł –Ľ–Ķ—á–Ķ–Ĺ–ł–ł –ĺ—Ā–Ľ–į–Ī–Ľ–Ķ–Ĺ–Ĺ—č—Ö –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ –Ņ—Ä–Ķ–ī–Ņ–ĺ—á—ā–Ķ–Ĺ–ł–Ķ —Ā–Ľ–Ķ–ī—É–Ķ—ā –ĺ—ā–ī–į–≤–į—ā—Ć –ľ–ĺ–Ĺ–ĺ—ā–Ķ—Ä–į–Ņ–ł–ł. –•–ł–ľ–ł–ĺ—ā–Ķ—Ä–į–Ņ–Ķ–≤—ā–ł—á–Ķ—Ā–ļ–ł–Ļ —ā—Ä–ł–Ņ–Ľ–Ķ—ā –Ņ—Ä–ĺ–ī–Ķ–ľ–ĺ–Ĺ—Ā—ā—Ä–ł—Ä–ĺ–≤–į–Ľ –Ņ—Ä–Ķ–ł–ľ—É—Č–Ķ—Ā—ā–≤–į –Ņ–ĺ –≤—Ā–Ķ–ľ –Ņ–ĺ–ļ–į–∑–į—ā–Ķ–Ľ—Ź–ľ –≤¬†—Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł–ł —Ā¬†–ľ–ĺ–Ĺ–ĺ—ā–Ķ—Ä–į–Ņ–ł–Ķ–Ļ –≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ–ĺ–ľ, –ĺ–ī–Ĺ–į–ļ–ĺ —ā—Ä–Ķ–Ī—É—é—ā—Ā—Ź –Ņ—Ä–ĺ–≤–Ķ—Ä–ĺ—á–Ĺ—č–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź. –ė–∑ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤ —Ü–Ķ–Ľ–Ķ–≤–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł –ī–Ľ—Ź –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź –†–ü–Ė –≤¬†–Ĺ–į—Ā—ā–ĺ—Ź—Č–Ķ–Ķ –≤—Ä–Ķ–ľ—Ź –ł—Ā–Ņ–ĺ–Ľ—Ć–∑—É–Ķ—ā—Ā—Ź —ā–ĺ–Ľ—Ć–ļ–ĺ —ć—Ä–Ľ–ĺ—ā–ł–Ĺ–ł–Ī. –ö–ĺ–ľ–Ī–ł–Ĺ–į—Ü–ł—Ź –≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ–į —Ā¬†—ć—Ä–Ľ–ĺ—ā–ł–Ĺ–ł–Ī–ĺ–ľ –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ¬†–Ĺ–Ķ–∑–Ĺ–į—á–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–ľ—É —É–≤–Ķ–Ľ–ł—á–Ķ–Ĺ–ł—é —Ā—Ä–ĺ–ļ–ĺ–≤ –∂–ł–∑–Ĺ–ł, –Ĺ–ĺ –Ņ—Ä–ł —ć—ā–ĺ–ľ —Ā—É—Č–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ –≤–ĺ–∑—Ä–į—Ā—ā–į–Ķ—ā —Ā—ā–ĺ–ł–ľ–ĺ—Ā—ā—Ć –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź –ł¬†—É—Ā–ł–Ľ–ł–≤–į–Ķ—ā—Ā—Ź —ā–ĺ–ļ—Ā–ł—á–Ĺ–ĺ—Ā—ā—Ć.
–ě—ā—Ä–ł—Ü–į—ā–Ķ–Ľ—Ć–Ĺ—č–Ķ —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā—č –Ņ—Ä–ł–≤–Ķ–ī–Ķ–Ĺ–Ĺ—č—Ö –≤—č—ą–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ –Ņ–ĺ –ļ–ĺ–ľ–Ī–ł–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–ł—é –≥–Ķ–ľ—Ü–ł—ā–į–Ī–ł–Ĺ–į —Ā¬†—ā–į—Ä–≥–Ķ—ā–Ĺ—č–ľ–ł –į–≥–Ķ–Ĺ—ā–į–ľ–ł –Ņ–ĺ–ī—ā–≤–Ķ—Ä–∂–ī–į—é—ā —ā–Ķ–ĺ—Ä–ł—é –ĺ¬†—Ā–Ľ–ĺ–∂–Ĺ–ĺ—Ā—ā–ł –ļ–į–Ĺ—Ü–Ķ—Ä–ĺ–≥–Ķ–Ĺ–Ķ–∑–į –†–ü–Ė. –ü–ĺ-–≤–ł–ī–ł–ľ–ĺ–ľ—É, –ī–Ľ—Ź —Ä–Ķ–į–Ľ–ł–∑–į—Ü–ł–ł –Ņ—Ä–ĺ—ā–ł–≤–ĺ–ĺ–Ņ—É—Ö–ĺ–Ľ–Ķ–≤–ĺ–≥–ĺ —ć—Ą—Ą–Ķ–ļ—ā–į –Ĺ–Ķ–ī–ĺ—Ā—ā–į—ā–ĺ—á–Ĺ–ĺ –Ņ–ĺ–ī–į–≤–Ľ–Ķ–Ĺ–ł—Ź —ā–ĺ–Ľ—Ć–ļ–ĺ –ĺ–ī–Ĺ–ĺ–≥–ĺ –∑–≤–Ķ–Ĺ–į –Ņ–į—ā–ĺ–≥–Ķ–Ĺ–Ķ—ā–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –Ņ—É—ā–ł. –Ě–Ķ–ĺ–Ī—Ö–ĺ–ī–ł–ľ–ĺ —Ä–į–∑—Ä–į–Ī–į—ā—č–≤–į—ā—Ć –ľ—É–Ľ—Ć—ā–ł–ľ–ĺ–ī–į–Ľ—Ć–Ĺ—č–Ļ –Ņ–ĺ–ī—Ö–ĺ–ī –ļ¬†–Ľ–Ķ—á–Ķ–Ĺ–ł—é —ć—ā–ĺ–≥–ĺ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź. –Ě–į–ł–Ī–ĺ–Ľ–Ķ–Ķ –Ņ–Ķ—Ä—Ā–Ņ–Ķ–ļ—ā–ł–≤–Ĺ—č–ľ –Ĺ–į–Ņ—Ä–į–≤–Ľ–Ķ–Ĺ–ł–Ķ–ľ —Ü–Ķ–Ľ–Ķ–≤–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł –†–ü–Ė —Ā–Ľ–Ķ–ī—É–Ķ—ā —Ā—á–ł—ā–į—ā—Ć –ł–∑—É—á–Ķ–Ĺ–ł–Ķ —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł PARP-–ł–Ĺ–≥–ł–Ī–ł—ā–ĺ—Ä–ĺ–≤, –ł–Ĺ–≥–ł–Ī–ł—ā–ĺ—Ä–ĺ–≤ —Ā–ł–≥–Ĺ–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –Ņ—É—ā–ł PI3K-Akt, –≤—č—Ź–≤–Ľ–Ķ–Ĺ–ł–Ķ –ľ–ł—ą–Ķ–Ĺ–Ķ–Ļ –ł¬†–į–Ĺ—ā–į–≥–ĺ–Ĺ–ł—Ā—ā–ĺ–≤ –ļ¬†–Ĺ–ł–ľ –≤¬†–ľ–ł–ļ—Ä–ĺ–ĺ–ļ—Ä—É–∂–Ķ–Ĺ–ł–ł –ĺ–Ņ—É—Ö–ĺ–Ľ–ł.
–£–≤–į–∂–į–Ķ–ľ—č–Ļ –Ņ–ĺ—Ā–Ķ—ā–ł—ā–Ķ–Ľ—Ć uMEDp!
–£–≤–Ķ–ī–ĺ–ľ–Ľ—Ź–Ķ–ľ –í–į—Ā –ĺ —ā–ĺ–ľ, —á—ā–ĺ –∑–ī–Ķ—Ā—Ć —Ā–ĺ–ī–Ķ—Ä–∂–ł—ā—Ā—Ź –ł–Ĺ—Ą–ĺ—Ä–ľ–į—Ü–ł—Ź, –Ņ—Ä–Ķ–ī–Ĺ–į–∑–Ĺ–į—á–Ķ–Ĺ–Ĺ–į—Ź –ł—Ā–ļ–Ľ—é—á–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ –ī–Ľ—Ź —Ā–Ņ–Ķ—Ü–ł–į–Ľ–ł—Ā—ā–ĺ–≤ –∑–ī—Ä–į–≤–ĺ–ĺ—Ö—Ä–į–Ĺ–Ķ–Ĺ–ł—Ź.
–ē—Ā–Ľ–ł –í—č –Ĺ–Ķ —Ź–≤–Ľ—Ź–Ķ—ā–Ķ—Ā—Ć —Ā–Ņ–Ķ—Ü–ł–į–Ľ–ł—Ā—ā–ĺ–ľ –∑–ī—Ä–į–≤–ĺ–ĺ—Ö—Ä–į–Ĺ–Ķ–Ĺ–ł—Ź, –į–ī–ľ–ł–Ĺ–ł—Ā—ā—Ä–į—Ü–ł—Ź –Ĺ–Ķ –Ĺ–Ķ—Ā–Ķ—ā –ĺ—ā–≤–Ķ—ā—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ—Ā—ā–ł –∑–į –≤–ĺ–∑–ľ–ĺ–∂–Ĺ—č–Ķ –ĺ—ā—Ä–ł—Ü–į—ā–Ķ–Ľ—Ć–Ĺ—č–Ķ –Ņ–ĺ—Ā–Ľ–Ķ–ī—Ā—ā–≤–ł—Ź, –≤–ĺ–∑–Ĺ–ł–ļ—ą–ł–Ķ –≤ —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā–Ķ —Ā–į–ľ–ĺ—Ā—ā–ĺ—Ź—ā–Ķ–Ľ—Ć–Ĺ–ĺ–≥–ĺ –ł—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į–Ĺ–ł—Ź –í–į–ľ–ł –ł–Ĺ—Ą–ĺ—Ä–ľ–į—Ü–ł–ł —Ā –Ņ–ĺ—Ä—ā–į–Ľ–į –Ī–Ķ–∑ –Ņ—Ä–Ķ–ī–≤–į—Ä–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ļ –ļ–ĺ–Ĺ—Ā—É–Ľ—Ć—ā–į—Ü–ł–ł —Ā –≤—Ä–į—á–ĺ–ľ.
–Ě–į–∂–ł–ľ–į—Ź –Ĺ–į –ļ–Ĺ–ĺ–Ņ–ļ—É ¬ę–í–ĺ–Ļ—ā–ł¬Ľ, –í—č –Ņ–ĺ–ī—ā–≤–Ķ—Ä–∂–ī–į–Ķ—ā–Ķ, —á—ā–ĺ —Ź–≤–Ľ—Ź–Ķ—ā–Ķ—Ā—Ć –≤—Ä–į—á–ĺ–ľ –ł–Ľ–ł —Ā—ā—É–ī–Ķ–Ĺ—ā–ĺ–ľ –ľ–Ķ–ī–ł—Ü–ł–Ĺ—Ā–ļ–ĺ–≥–ĺ –≤—É–∑–į.