Диабетическая полиневропатия: от теории к практике
- Аннотация
- Статья
- Ссылки
- English


Сахарный диабет (СД) становится одной из самых серьезных проблем в сфере здравоохранения в ХХI в. Предполагается, что число людей, страдающих СД, к 2030 г. увеличится в полтора раза по сравнению с 2011 г. и достигнет пандемического уровня в 522 млн (9,9% населения планеты) [1].
Диабетическая полиневропатия (ДПН) – наиболее частое осложнение СД, приводящее к формированию диабетической стопы, нарушению ходьбы, падениям. Приблизительно 20–30% пациентов с ДПН страдают от невропатической боли [2]. ДПН значительно снижает качество жизни пациентов с СД и существенно увеличивает стоимость лечения.
Эпидемиология
По данным различных авторов, распространенность ДПН варьирует от 50% при клиническом обследовании до 90–100% при проведении электромиографического обследования у пациентов с длительностью сахарного диабета более 20 лет [3, 4]. У 8% пациентов с впервые установленным диагнозом СД уже находят клинические признаки ДПН [5]. У 5% пациентов неврологические нарушения – это первые признаки заболевания и основания для исследования углеводного обмена [6]. Распространенность ДПН у пациентов с СД 1 типа выше (54–59%), чем у пациентов с СД 2 типа (45%) [7].
Патогенез
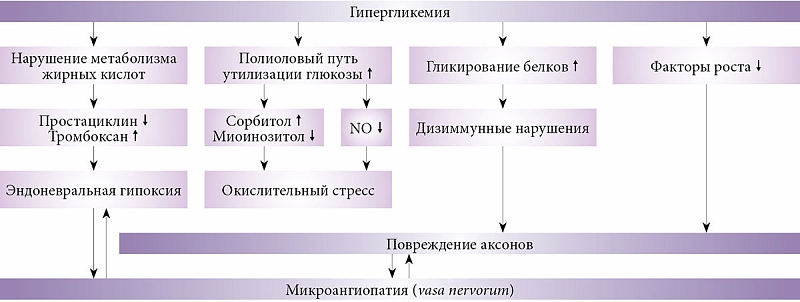
Патогенез ДПН сложен: периферические нервы при сахарном диабете страдают в результате как дисметаболических, так и сосудистых нарушений (рис. 1). Периферические нейроны потребляют глюкозу инсулинонезависимо посредством пассивной диффузии, что в условиях гипергликемии приводит к чрезмерной активации полиолового пути окисления глюкозы. Поток глюкозы по полиоловому пути в различных тканях увеличивается на 11–33%. Происходит накопление сорбитола и фруктозы. Следствием избытка сорбитола в клетке становится осмотическое напряжение, что в конечном итоге вызывает повреждение нейрона. Снижается активность ферментов, участвующих в проведении нервного импульса по волокну (Na+/K+-АТФаза). Гипергликемия усиливает процессы неферментативного и ферментативного гликирования структурных белков нервного волокна (миелина и тубулина). В результате образуются конечные продукты гликирования белков, которые нарушают функциональную активность нейрона и стимулируют синтез провоспалительных цитокинов [8–10].
Еще один механизм дегенерации нейрона в условиях гипергликемии – окислительный стресс, который подразумевает снижение уровня антиоксидантов и накопление активных форм кислорода (свободных радикалов).
Перекисное окисление липидов приводит к нарушению структуры мембраны нейрона и в конечном итоге к апоптозу нейронов и глиальных клеток. Окислительный стресс развивается не только в нейронах, но и в сосудах, питающих нейроны (vasa nervorum и vasa vasorum). Активация перекисного окисления липидов способствует эндотелиальной дисфункции, которая в свою очередь вызывает ишемическое повреждение нейронов (микротромбозы и окклюзии капилляров) и нарушает выработку факторов роста в нервной ткани, снижая тем самым регенеративный потенциал нейрона [11–13]. Следствием активации протеинкиназы С на одной из стадий окислительного стресса становится уменьшение образования оксида азота (NO), что также ведет к эндоневральной гипоксии. Недостаточная активность антиоксидантных ферментов при СД определяется генетическими факторами, что подтверждается изучением полиморфизма генов таких ферментов антиоксидантной системы организма, как каталаза (при диабетической ретинопатии) и супероксиддисмутаза (при диабетической полиневропатии) [11].
В условиях гипергликемии повреждаются не только нейроны и эндотелиальные клетки сосудов, но и глиальные клетки, влияющие на синаптическую связь между нейронами. В результате повреждения клеток глии высвобождаются провоспалительные цитокины: интерлейкин 1-бета, интерлейкин 16, фактор некроза опухоли альфа. Действие этих цитокинов лежит в основе патогенеза аллодинии и гипералгезии.
Хороший метаболический контроль при СД 1 типа позволяет снизить вероятность развития ДПН на 60–70%, при СД 2 типа – всего лишь на 5–7% [14, 15]. Более того, несмотря на хороший метаболический контроль, у 40% пациентов с СД развивается ДПН. Пациенты с СД с уровнем HbA1C ниже 5,4% также иногда имеют клинические признаки ДПН [16]. Эти данные свидетельствуют об участии других неизученных факторов, повреждающих нейроны, и подтверждают необходимость дальнейшего изучения патогенеза ДПН.
Факторы риска
Выделяют модифицируемые и немодифицируемые факторы риска развития ДПН. Главный модифицируемый фактор риска ДПН – гипергликемия, что было доказано в крупных проспективных исследованиях DCCT и UKPDS [17, 18]. К модифицируемым факторам риска ДПН также относятся злоупотребление алкоголем, курение, артериальная гипертензия, повышенный уровень триглицеридов, индекса массы тела. Независимые факторы риска ДПН – пожилой возраст, мужской пол, наследственная отягощенность по полиневропатии, длительное течение сахарного диабета, APOE-генотип, гиперактивность гена альдозоредуктазы.
Известно, что к поражению периферических нервов приводит не только плохой метаболический контроль, но и быстрая коррекция гипергликемии. Выделена особая форма – невропатия, индуцированная лечением диабета. Это ятрогенная невропатия тонких волокон, которая клинически проявляется остро развивающейся невропатической болью и/или автономной дисфункцией в течение первых восьми недель агрессивного лечения СД. Под агрессивным лечением понимается снижение уровня HbA1C свыше 2% за три месяца терапии [19]. Иначе данный тип ДПН называют инсулиновым невритом [20]. Многие годы считалось, что инсулиновый неврит – редкая причина острой диабетической невропатии, однако последние публикации говорят об обратном. Согласно исследованиям C.H. Gibbons и R. Freeman, у 10,9% пациентов с СД обнаруживается индуцированная лечением невропатия [19]. Считается, что данная форма чаще встречается у пациентов с СД 1 типа, хотя бывает и среди пациентов с СД 2 типа, получающих инсулин. Патогенез данного вида периферической невропатии изучен недостаточно. Предполагается, что быстрое снижение уровня глюкозы приводит к гемодинамическим изменениям (артериовенозный сброс), что вызывает эндоневральную гипоксию тонких волокон [21].
Клинические особенности
Симптомы ДПН значительно варьируются. В одних случаях возникают нарушения чувствительности и невропатическая боль, в других – автономная дисфункция, в третьих – мышечная слабость. 90% пациентов с ДПН имеют симметричную дистальную сенсорную полиневропатию (СДСП). СДСП обычно начинается исподволь и медленно прогрессирует. В клинической картине доминируют позитивные и негативные симптомы симметричного поражения чувствительных нервов.
Позитивные симптомы – ощущение жжения, покалывания, режущая, колющая боль, боль по типу прохождения электрического тока, аллодиния (возникновение болевого ощущения в ответ на неболевой стимул, например, при прикосновении одежды). Боль чаще возникает в ночные часы и ослабевает при ходьбе, что отличает ее от боли при поражении сосудов нижних конечностей. Позитивные симптомы чаще связаны с поражением тонких немиелинизированных волокон (типа С), возникают из-за дизингибиции сенсорного ответа поврежденного волокна [22].
К негативным симптомам относятся чувство онемения, ощущение «мертвой» или «уснувшей» стянутой конечности, выпадение сухожильных рефлексов, сенситивная атаксия. Негативные симптомы связаны со снижением скорости или отсутствием проведения импульса по нервному волокну, чаще по толстому миелинизированному волокну (типа А). Пациенты с негативными симптомами относятся к группе риска по развитию диабетической стопы из-за отсутствия защитной функции болевой и тактильной чувствительности [21, 22].
Чувствительные симптомы обычно вначале появляются в пальцах и постепенно поднимаются в проксимальном направлении, вовлекая стопы, голени и бедра. Это связано с тем, что в первую очередь страдают длинные аксоны. Данный вид невропатии часто называют зависимым от длины [24, 25]. В конечном итоге прогрессирование СДСП приводит к развитию синдрома диабетической стопы с формированием остеоартропатии в виде «сустава Шарко», образованием язвенных дефектов в местах максимального давления в стопе, последующим присоединением вторичной инфекции, развитием гангрены и в финале ампутацией конечности.
Моторные симптомы, такие как мышечная слабость и атрофия, как правило, выражены минимально и чаще возникают при значительной длительности заболевания [26]. Выраженная сенсорная атаксия не характерна для ДПН, поэтому при ее выявлении необходимо исключать другие причины [27].
Более половины пациентов с СДСП не знают о наличии у себя этого осложнения. С одной стороны, если пациент имеет диабетическую нефропатию и ретинопатию, то в большинстве случаев этот пациент страдает и СДСП. С другой стороны, 25–62% пациентов с идиопатической периферической полиневропатией имеют предиабет. У 11–25% из них отмечается периферическая сенсорная невропатия, а у 13–21% – невропатическая боль [23].
Автономные расстройства часто сочетаются с СДСП. При этом в клинической картине доминируют признаки дисфункции сердечно-сосудистой системы (ортостатическая дисфункция, фиксированный пульс), желудочно-кишечного тракта (запоры, поносы, гастропарез), мочеполовой системы (импотенция, ретроградная эякуляция, нейрогенный мочевой пузырь). Автономная невропатия может приводить к безболевым инфарктам миокарда, злокачественным аритмиям, внезапной смерти. Сердечно-сосудистая форма автономной невропатии в три раза увеличивает смертность пациентов с СД [28]. Денервация надпочечников может быть причиной уменьшения автономной реакции на гипогликемию, что затрудняет ее диагностику и лечение.
В соответствии с часто используемой классификацией P. Dyck и соавт., выделяют три стадии ДПН: легкую, умеренно выраженную, тяжелую (табл. 1) [29].
К редким формам поражения периферической нервной системы при СД относятся:
- острая диабетическая офтальмоплегия: одностороннее поражение III, реже IV и VI пар краниальных нервов;
- острая полиневропатия конечностей или туловища, включающая болевую торакоабдоминальную невропатию, диабетическую радикулопатию;
- симметричная проксимальная моторная полиневропатия;
- асимметричная проксимальная моторная полиневропатия (диабетическая амиотрофия);
- множественная мононевропатия.
Эти виды невропатий чаще возникают остро или подостро, в дебюте сопровождаются выраженным болевым синдромом. В патогенезе этих видов невропатий ведущим механизмом являются не столько нарушения метаболизма глюкозы, как при хронических формах, сколько ишемическое повреждение нервов – инфаркты нервов [30].
Отдельно стоит упомянуть разновидность диабетической мононевропатии – туннельную невропатию. Патогенез поражения периферического нерва связан с компрессией его в анатомически узком канале. Почти у трети пациентов с ДПН наблюдаются клинические проявления СДСП и туннельной невропатии срединных нервов на уровне карпальных каналов [23].
СД часто сочетается с дефицитом витамина В12, уремией, гипотиреозом, алкоголизмом, ревматическими заболеваниями. Эти патологические состояния также могут приводить к полиневропатии. В клинической практике встречаются ятрогенные причины недостатка витамина В12, связанные с терапией метформином [30].
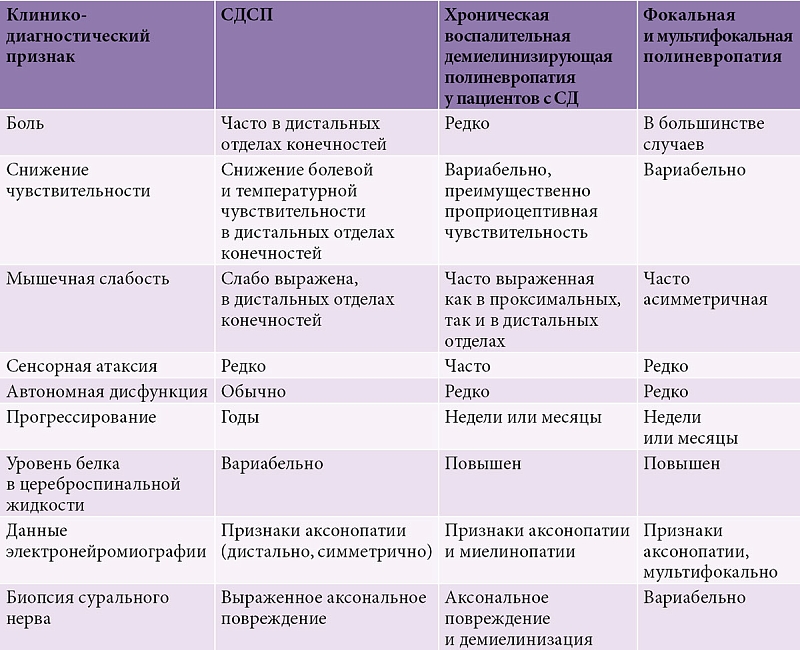
Хроническая воспалительная демиелинизирующая полиневропатия несколько чаще встречается у пациентов с СД, чем в популяции. Поэтому ДПН следует диагностировать только в том случае, когда исключены другие заболевания, способные вызывать сходную симптоматику (табл. 2).
Диагностика
Диагностика ДПН базируется в первую очередь на клинических данных, анамнезе, характерных жалобах, полиневропатическом типе чувствительных и двигательных расстройств. Ведущие мировые эксперты по заболеваниям периферической нервной системы P.B. Dyck и P.J. Dyck (1999) выделяют следующие диагностические критерии ДПН [29]:
- сахарный диабет;
- продолжительная хроническая гипергликемия;
- дистальная симметричная сенсомоторная полиневропатия;
- исключение других причин сенсомоторной полиневропатии;
- диабетическая ретино- или нефропатия близки по тяжести к полиневропатии.
Для подтверждения диагноза ДПН в некоторых случаях используют электромиографию и исследование соматосенсорных вызванных потенциалов. При электромиографии обнаруживают признаки как аксонопатии (снижение амплитуды М-ответа), так и демиелинизации (легкое или умеренное снижение скорости проведения по нервам, увеличение дистальной латенции, изменение показателей F-волн). В редких случаях для верификации диагноза ДПН используется биопсия сурального нерва. Патоморфологический субстрат ДПН – истончение миелинизированных волокон, диффузные или локальные демиелинизированные участки, дегенерация аксонов, уменьшение просвета vasa nervorum и утолщение базальной мембраны капилляров [31].
Все больные СД, независимо от характерных жалоб, должны проходить ежегодный скрининг на полиневропатию (рис. 2):
- исследование болевой чувствительности;
- исследование тактильной чувствительности с помощью монофиламента;
- исследование вибрационной чувствительности с помощью градуированного камертона.
Для диагностики автономной недостаточности проводятся кардиоваскулярные тесты, отслеживаются изменение пульса при пробе Вальсальвы и глубоком вдохе после вставания из положения лежа, изменение артериального давления в тесте на сжимание рук и ортостатической пробе. Наиболее чувствительный и простой тест – исследование дыхательной аритмии. В процессе регистрации электрокардиограммы больной глубоко вдыхает шесть раз за минуту, при этом продолжительность вдоха и выдоха должна составлять пять секунд. В норме разница между максимальной (вдох) и минимальной (выдох) частотой сердечных сокращений должна быть не менее 10. Этот тест рекомендуется проводить у больных с СД с интервалом в один-два года [30].
Лечение и профилактика
Тщательный контроль гликемии в дебюте СД позволяет отсрочить клиническую манифестацию полиневропатии почти на два года. Уже имеющееся поражение периферических нервов может быть приостановлено и даже подвергнуться обратному развитию при достижении надлежащего метаболического контроля. Однако наиболее нагляден этот эффект при СД 1 типа, в то время как у пациентов с СД 2 типа достижение надлежащего метаболического контроля не всегда приводит к регрессу невропатических расстройств [32].
Немаловажно в лечении и профилактике ДПН контролировать коморбидную патологию, усугубляющую клинические проявления невропатии: артериальную гипертензию, дислипидемию, избыточную массу тела [33].
Эффективность препаратов первой линии в лечении невропатической боли при ДПН (трициклических антидепрессантов – амитриптилина, нортриптилина; ингибиторов обратного захвата серотонина и норадреналина – дулоксетина, венлафаксина; антиконвульсантов – габапентина, прегабалина) сопоставима [34, 35]. В США одобрены к применению при неврологической боли, связанной с ДПН, только венлафаксин и прегабалин [36].
Последние годы активно изучают средства патогенетической терапии ДПН, такие как ингибиторы альдозоредуктазы, протеинкиназы С, «чистильщики» свободных радикалов [37]. Несмотря на многообещающие доклинические результаты, эффективность ни одного из этих препаратов на сегодняшний день не доказана.
Учитывая важность окислительного стресса в патогенезе ДПН, оправданно использовать в терапии данного осложнения антиоксиданты, например альфа-липоевую (тиоктовую) кислоту (АЛК). АЛК была открыта в 1948 г., в 1951 г. ее выделили из экстрактов говяжьей печени и определили ее структурную формулу. Клинические исследования эффективности АЛК при различных заболеваниях были начаты еще в 1950-х гг. [38]. Антиоксидантный эффект АЛК обусловлен наличием двух тиоловых групп в молекуле, благодаря чему она может связывать свободные радикалы и ионы металлов, входящие в состав ферментов, катализирующих процессы перекисного окисления липидов. АЛК хорошо растворяется как в водной, так и в жировой среде и легко проникает через клеточные мембраны. Противодействуя окислительному стрессу, АЛК улучшает микроциркуляцию в эндонервии, снижает риск ишемического поражения нервной ткани и улучшает функциональную активность нейронов. Кроме антиоксидантного, доказаны и другие метаболические эффекты АЛК: усиление процесса утилизации глюкозы, повышение активности цикла Кребса, снижение периферической резистентности к инсулину. Экспериментальные данные позволяют предположить и нейротрофическое действие АЛК, заключающееся в стимуляции продукции фактора роста и регенерации аксонов, нормализации аксонального транспорта [39, 40].
В многочисленных клинических исследованиях были продемонстрированы высокая эффективность и хорошая переносимость АЛК при ДПН. Так, исследование ALADIN показало, что наиболее эффективной и безопасной дозой при инфузионном введении АЛК была доза 600 мг/сут. При увеличении дозы до 1200 мг/сут чаще наблюдались побочные эффекты [41].
В исследовании ALADIN II оценивалась длительная терапия АЛК. Препарат назначался 65 пациентам сначала внутривенно в течение первых пяти дней, а затем перорально по 600 (одна группа) и 1200 мг/сут (другая группа) в течение двух лет. Эффективность терапии оценивали с помощью количественных шкал. Через 24 месяца терапии было получено статистически значимое (p < 0,05) улучшение показателей количественных тестов в обеих контрольных группах по сравнению с группой, получавшей плацебо [42].
В рандомизированном двойном слепом плацебоконтролируемом исследовании SYDNEY в 2001 г. приняли участие 120 больных СД 1 и 2 типа из США и России. 60 человек получали плацебо, а 60 – АЛК. Прием АЛК положительно влиял на клинические проявления ДПН, электрофизиологические показатели (данные электронейромиографии) и автономную дисфункцию [43].
В исследовании SYDNEY II была продемонстрирована связь между дозой АЛК и скоростью наступления терапевтического эффекта. При назначении 600 мг/сут улучшение самочувствия отмечалось через три недели лечения, а при использовании более высоких доз (1200 и 1800 мг/сут) – в более короткие сроки [44, 45].
В исследовании DEKAN было установлено, что на фоне четырехмесячного приема АЛК 800 мг/сут перорально у пациентов с СД 2 типа отмечалось ослабление проявлений автономной дисфункции [46].
На основе результатов клинических исследований рекомендуется следующая схема терапии ДПН препаратом АЛК: начальная инфузионная терапия 300–600 мг/сут в/в капельно в течение двух – четырех недель с последующим переходом на пероральный прием 600 мг/сут в течение трех-четырех месяцев [31].
Заключение
ДПН – одно из наиболее распространенных осложнений CД, которое встречается более чем у 50% пациентов. Риск развития ДПН пропорционален степени и длительности гипергликемии. Кроме тщательного метаболического контроля не существует доказанных методов профилактики данной патологии. Важно как можно раньше выявить ДПН. Симптоматическое лечение проводится при болевых формах невропатии и заключается в использовании антиконвульсантов и антидепрессантов. Патогенетическая терапия подразумевает использование АЛК. Многочисленные данные, полученные в ходе многоцентровых рандомизированных исследований, свидетельствуют о высокой эффективности и безопасности применения препаратов АЛК, что делает их незаменимыми в лечении ДПН.
V.B. Sosina, V.V. Zakharov
City Clinical Hospital № 51, Moscow
I.M. Sechenov First Moscow State Medical University
Contact person: Vladimir Vladimirovich Zakharov, zakharovenator@gmail.com
Diabetic neuropathy (DN) is a most common long-term complication of diabetes, which leads to poor quality of life and disability. This article presents literature data on the prevalence, pathogenesis, clinical features and predictors of DN. The recommendations for the prevention and treatment of DN are presented in accordance with the results of clinical studies in this area. The important role of antioxidants, including alpha-lipoic acid in the treatment of DN is shown.
Уважаемый посетитель uMEDp!
Уведомляем Вас о том, что здесь содержится информация, предназначенная исключительно для специалистов здравоохранения.
Если Вы не являетесь специалистом здравоохранения, администрация не несет ответственности за возможные отрицательные последствия, возникшие в результате самостоятельного использования Вами информации с портала без предварительной консультации с врачом.
Нажимая на кнопку «Войти», Вы подтверждаете, что являетесь врачом или студентом медицинского вуза.