Взаимосвязь болезни Паркинсона и сахарного диабета 2 типа
- Аннотация
- Статья
- Ссылки
- English
Установлено, что сахарный диабет 2 типа ассоциируется с повышением риска развития болезни Паркинсона, скоростью ее прогрессирования, тяжестью моторного и когнитивного дефицита.
Одним из ключевых звеньев патогенеза сахарного диабета 2 типа является инсулинорезистентность. Процесс, аналогичный периферической инсулинорезистентности, происходит в тканях головного мозга пациентов с болезнью Паркинсона.
На сегодняшний день не существует эффективных методов лечения болезни Паркинсона, которые бы изменили ее течение. Применение противодиабетических препаратов в качестве нейропротективных средств открывает новые возможности таргетной терапии патологии.
В статье рассматриваются эпидемиологические, клинические и патофизиологические аспекты связи этих двух заболеваний, а также возможности перепрофилирования противодиабетических препаратов для лечения болезни Паркинсона.
Установлено, что сахарный диабет 2 типа ассоциируется с повышением риска развития болезни Паркинсона, скоростью ее прогрессирования, тяжестью моторного и когнитивного дефицита.
Одним из ключевых звеньев патогенеза сахарного диабета 2 типа является инсулинорезистентность. Процесс, аналогичный периферической инсулинорезистентности, происходит в тканях головного мозга пациентов с болезнью Паркинсона.
На сегодняшний день не существует эффективных методов лечения болезни Паркинсона, которые бы изменили ее течение. Применение противодиабетических препаратов в качестве нейропротективных средств открывает новые возможности таргетной терапии патологии.
В статье рассматриваются эпидемиологические, клинические и патофизиологические аспекты связи этих двух заболеваний, а также возможности перепрофилирования противодиабетических препаратов для лечения болезни Паркинсона.
Введение
Болезнь Паркинсона является наиболее частым двигательным расстройством, вторым по распространенности нейродегенеративным [1] и самым быстро распространяющимся неврологическим заболеванием. Так, количество страдающих болезнью Паркинсона с 1990 по 2016 г. увеличилось более чем вдвое, при этом ожидается, что эта тенденция будет сохраняться в течение следующих 30 лет [2].
Несмотря на многолетние исследования, этиология болезни Паркинсона остается неизвестной. От 5 до 10% случаев обусловлены наследственной предрасположенностью. В большинстве случаев заболевание является спорадическим и имеет мультифакторную природу [3].
Ключевым молекулярным событием в развитии нейродегенерации при болезни Паркинсона считается нарушение конформации везикулярного белка α-синуклеина с формированием нейротоксичных цитоплазматических агрегатов и телец/нейритов Леви [3].
Классические двигательные проявления патологии, такие как гипокинезия, ригидность, тремор покоя, нарушение ходьбы, обусловлены преимущественно утратой нигростриарных дофаминергических нейронов. Спектр немоторных симптомов более широк и часто связан не только с дегенерацией нигростриарных нейронов и дефицитом дофамина в головном мозге, но и с другими механизмами, включая нейроэндокринные и нейрометаболические [4].
Сахарный диабет – одно из наиболее распространенных хронических метаболических заболеваний [5].
В многочисленных исследованиях была выявлена связь между болезнью Паркинсона и сахарным диабетом 2 типа [6–11].
В статье рассматриваются эпидемиология, патогенез, а также общие подходы к терапии сахарного диабета 2 типа и болезни Паркинсона.
Эпидемиология
Диабет является глобальной проблемой здравоохранения. Данной патологией страдают почти 537 млн человек во всем мире [12].
Согласно эпидемиологическим данным, сахарный диабет 2 типа увеличивает риск возникновения болезни Паркинсона [13, 14]. Развитие сахарного диабета 2 типа у пациентов с болезнью Паркинсона коррелирует с быстрым прогрессированием заболевания, а также тяжестью моторного и когнитивного дефицита [14, 15]. Однако эти данные находят подтверждение не во всех работах [16, 17].
Патогенез
Инсулинорезистентность является одним из ключевых звеньев патогенеза как сахарного диабета 2 типа, так и болезни Паркинсона. Для обеих патологий характерны аберрантное накопление белка, лизосомная и митохондриальная дисфункция, хроническое системное воспаление [6, 18].
Между инсулином и дофамином возможна функциональная связь. Так, на сегодняшний день убедительно доказано действие дофамина на β-клетки поджелудочной железы, а также плейотропные эффекты инсулина в центральной нервной системе, в значительной степени независимые от утилизации глюкозы [19].
В недавних работах установлено взаимодействие патологических форм белка амилина и α-синуклеина [20], что приводило к их коагрегации как in vitro, так и in vivo. Это позволяет предположить как роль α-синуклеина в образовании амилоида β-клеток, так и возможную роль амилина в формировании амилоидов α-синуклеина в клетках поджелудочной железы [21–23].
Для диабета и болезни Паркинсона характерны общие патологические микрососудистые изменения в головном мозге. В исследовании O.F. Elabi и соавт. по оценке влияния 21-недельного воздействия диеты с высоким содержанием жиров в сочетании с индуцированной токсинами моделью болезни Паркинсона на микрососудистые изменения установлено, что это приводило к значительному истощению перицитов, отсутствию ангиогенного ответа и значительному снижению взаимодействия микроглии/сосудов, что указывало на усугубление сосудистой патологии [24].
Открытие общих патофизиологических характеристик диабетической ретинопатии и болезни Паркинсона позволило предположить наличие положительной связи между этими патологиями.
До недавнего времени диабетическая ретинопатия считалась преимущественно микрососудистым заболеванием. Однако последние данные идентифицируют нейродегенерацию сетчатки как раннее событие ее патогенеза.
Болезнь Паркинсона и диабетическая ретинопатия коррелируют по заболеваемости, клиническим проявлениям и патофизиологическим механизмам, обусловленным нарушением активности дофамина (снижение уровня дофамина, повышение экспрессии α-синуклеина и аномальная экспрессия нейротрофических факторов) [25]. Однако доказательств того, что диабетическая ретинопатия относится к независимым факторам риска развития болезни Паркинсона, не обнаружено [26].
Накапливаются данные о том, что конечные продукты усиленного гликирования и их предшественник метилглиоксаль также влияют на изменение дофаминергической системы.
В исследовании, проведенном Y.Q. Lv и соавт., показано, что нейровоспаление, вызванное гипергликемией, способствовало нигростриарной дегенерации и прогрессированию болезни Паркинсона [27].
Однако E. de Pablo-Fernández и соавт. установили, что более быстрое прогрессирование болезни Паркинсона и повышение смертности среди пациентов, страдающих сахарным диабетом 2 типа, не связано с вкладом сосудистой патологии или усилением отложения α-синуклеина в ключевых областях мозга [28]. Исследователи предположили, что на течение болезни Паркинсона влияет дополнительная неспецифическая нейродегенерация, связанная с хронической инсулинорезистентностью мозга.
Инсулин играет важную роль в росте и регенерации нейронов, оказывает нейротрофическое, нейромодулирующее воздействие, способствует поддержанию нейропластичности [29–33]. Таким образом, нарушение передачи сигналов инсулина в клетках головного мозга потенциально может как предрасполагать к возникновению, так и способствовать прогрессированию нейродегенерации.
Инсулинорезистентность – состояние, сопровождающееся нарушением биологического ответа периферических тканей на воздействие эндогенного или экзогенного инсулина. В широком смысле данное понятие отражает реакцию периферических тканей на все эффекты инсулина, включая его воздействие на жировой и белковый метаболизм. Однако клинический термин «инсулинорезистентность» характеризует это состояние как нарушение ответа тканей на сахароснижающее действие инсулина [34].
Периферическая инсулинорезистентность. Многочисленные исследования указывают на высокий уровень аномальной толерантности к глюкозе у пациентов с болезнью Паркинсона [35, 36]. В то же время некоторые авторы сообщают о том, что явно нарушенный метаболизм глюкозы встречался примерно у 20% больных [37].
В большей степени инсулинорезистентность характерна для пациентов с избыточным весом или ожирением, что нередко встречается на ранних стадиях болезни Паркинсона из-за снижения физической активности, побочных эффектов препаратов [38–41], а также после глубокой стимуляции головного мозга [42].
Однако E. Hogg и соавт. выявили высокий процент худых среди пациентов с болезнью Паркинсона, у которых имела место инсулинорезистентность [43]. Исследователи также установили, что почти две трети лиц с болезнью Паркинсона, но без сахарного диабета 2 типа, могут быть инсулинорезистентными, несмотря на нормальный уровень глюкозы в крови натощак и во многих случаях нормальный уровень гликированного гемоглобина (HbA1c).
Центральная инсулинорезистентность. Исторически считалось, что инсулин обладает исключительно периферическим действием. Однако в последнее десятилетие было установлено, что инсулин может преодолевать гематоэнцефалический барьер и влиять на клеточный и митохондриальный метаболизм не только на периферии, но и в головном мозге [29, 44–46]. В частности, показано, что процесс, аналогичный периферической инсулинорезистентности, происходит в головном мозге пациентов с болезнью Паркинсона [34].
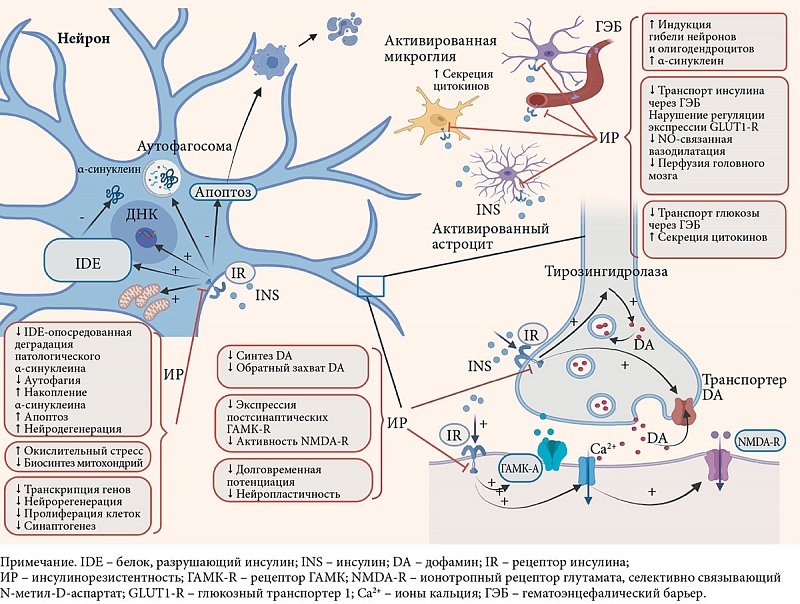
Существует тесная связь между центральной инсулинорезистентностью и нарушением функции гематоэнцефалического барьера при болезни Паркинсона [47, 48], модуляцией синаптической передачи [11, 18] и аутофагией [49], нарушением функции митохондрий и системным воспалением [50, 51] (рис. 1).
В связи со сказанным ранее особый интерес могут представлять результаты исследования M. Tagliati и соавт., которые установили, что инсулинорезистентность тканей головного мозга у пациентов с болезнью Паркинсона не зависела от периферической инсулинорезистентности [52].
Важно отметить, что она может быть как причиной [53], так и следствием нейродегенерации [54].
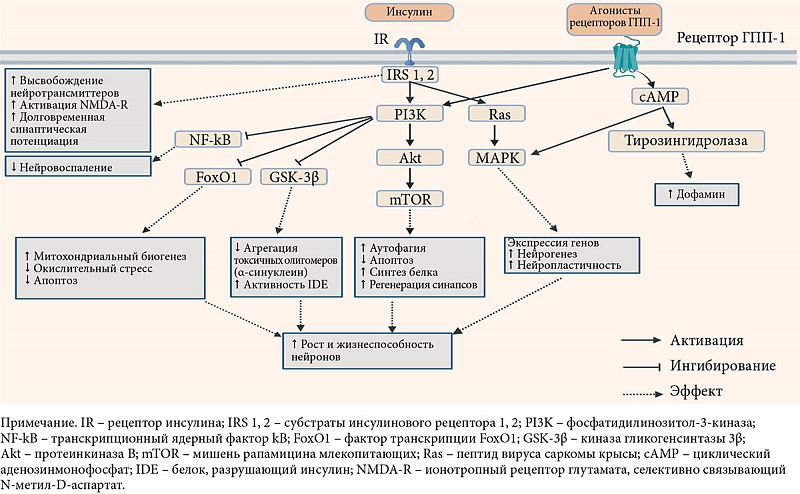
Нарушение клеточных механизмов передачи сигналов инсулина в головном мозге. Передача сигналов инсулина в нейронах происходит посредством двух сигнальных путей, известных как путь PI3K/Akt (комплекс протеинкиназы В) и путь митоген-активируемой протеинкиназы (MAPK).
Снижение передачи сигналов инсулина уменьшает активность Akt, модулируя активность множества киназ, включая механистическую мишень рапамицина, киназу гликогенсинтазы 3β и фактор транскрипции FoxO1, которые регулируют процессы, участвующие в развитии болезни Паркинсона [6], такие как деградация α-синуклеина [55, 56], митохондриальный биогенез и модуляция путей воспалительного и окислительного стресса [18, 33] (рис. 2).
Клинические особенности болезни Паркинсона на фоне сахарного диабета 2 типа
Влияние инсулинорезистентности в рамках сахарного диабета и метаболического синдрома на моторные проявления болезни Паркинсона
Согласно данным некоторых исследований, метаболический синдром и сахарный диабет коррелируют с тяжестью моторного дефицита [15, 37, 57]. При этом высокий уровень триглицеридов ассоциируется с защитным эффектом, высокое кровяное давление и низкий уровень холестерина липопротеинов высокой плотности – с ухудшением моторной функции и повышением риска падений [58]. Кроме того, в ряде исследований показано, что не только высокий, но и низкий уровень HbA1c может быть связан с ускорением прогрессирования двигательной симптоматики при болезни Паркинсона [59].
Диабет может способствовать постуральной нестабильности и затруднению походки через механизмы, не связанные с нигростриарной дофаминергической денервацией [14, 57, 60]. В то же время увеличение степени брадикинезии, скорее всего, обусловлено дофаминергической дисфункцией [61. В исследовании J.D. Meléndez-Flores и соавт. не было выявлено связи компонентов метаболического синдрома с моторными проявлениями болезни Паркинсона [62]. Однако, используя метаанализ традиционных обсервационных исследований и генетических данных, H. Chohan и соавт. [63] получили убедительные доказательства роли сахарного диабета 2 типа в более быстром прогрессировании двигательных нарушений у пациентов с болезнью Паркинсона.
Влияние инсулинорезистентности в рамках сахарного диабета и метаболического синдрома на немоторные проявления болезни Паркинсона
Корреляция метаболического синдрома и сахарного диабета 2 типа с риском возникновения когнитивных нарушений и тяжестью когнитивного дефицита у пациентов с болезнью Паркинсона неоднократно подтверждалась в разных работах [62, 63]. Так, M.R. Ashraghi и соавт. показали, что у пациентов с болезнью Паркинсона и сахарным диабетом 2 типа дефицит внимания и исполнительной функции был выражен намного сильнее, чем у лиц с болезнью Паркинсона, но без сахарного диабета [64].
В нескольких клинических и экспериментальных исследованиях установлена корреляция между когнитивными нарушениями и инсулинорезистентностью головного мозга [65]. Согласно данным C. Willmann и соавт. [66] и M.R. Ashraghi и соавт. [64], снижение когнитивных способностей у пациентов с инсулинорезистентностью преимущественно было обусловлено нарушением передачи сигналов инсулина в головном мозге, а не секрецией инсулина и уровнем гликемии.
Одним из наиболее важных открытий стала связь между патогенезом деменции, ассоциированной с болезнью Паркинсона, и механизмами развития инсулинорезистентности [67]. У лиц с деменцией вследствие болезни Паркинсона в два раза чаще встречалась инсулинорезистентность по сравнению с пациентами с болезнью Паркинсона, но без деменции [68].
При этом деменция со смешанным морфологическим вариантом (патология паркинсонического типа + патология альцгеймеровского типа) может быть особенно тесно связана с центральной инсулинорезистентностью [67].
Отмечено также влияние метаболического синдрома и сахарного диабета 2 типа на развитие депрессии [69, 70]. Однако при болезни Паркинсона получены лишь отдельные доказательства связи депрессии с сахарным диабетом 2 типа и инсулинорезистентностью [71].
Метаболический синдром и болезнь Паркинсона
Сахарный диабет 2 типа считается компонентом метаболического синдрома.
В исследовании дисбаланса глюкозы у пациентов с болезнью Паркинсона было предложено рассматривать метаболический синдром как новое немоторное последствие дизавтономии [38]. В то же время J. Roh и соавт. показали, что метаболический синдром увеличивает риск развития болезни Паркинсона прямо пропорционально увеличению количества компонентов метаболического синдрома [72]. В исследовании G. Nam и соавт. абдоминальное ожирение, высокое кровяное давление, низкий уровень липопротеинов высокой плотности, высокий уровень триглицеридов и глюкозы в крови натощак увеличивали частоту случаев развития болезни Паркинсона [73]. Наличие сразу пяти компонентов метаболического синдрома удваивало риск возникновения патологии [73]. В то же время в исследовании K. Sääksjärvi и соавт. высокий уровень триглицеридов в плазме крови ассоциировался со снижением риска развития болезни Паркинсона, особенно среди мужчин [74].
Общие терапевтические подходы
На сегодняшний день не существует клинически эффективных нейропротективных или модифицирующих течение болезни Паркинсона методов лечения.
С учетом влияния инсулинорезистентности на течение болезни Паркинсона представляют особый интерес способы восстановления чувствительности тканей к инсулину.
Известно, что низкокалорийная диета и физические упражнения, восстанавливающие чувствительность тканей к инсулину, положительно влияют на течение болезни Паркинсона. Не так давно было показано, что средиземноморская диета может играть нейропротективную роль, снижая риск развития патологии [75]. Необходимо отметить, что не во всех эпидемиологических исследованиях продемонстрировано положительное влияние средиземноморской диеты при нейродегенеративных заболеваниях [76].
Получены также данные о дозозависимой пользе упражнений при болезни Паркинсона, о способности упражнений высокой интенсивности нормализовать кортикомоторную возбудимость на ранних стадиях заболевания [77].
Актуальным направлением современных исследований является оценка возможностей коррекции инсулинорезистентности сахароснижающими препаратами.
Инсулин
С учетом описанных выше нейрозащитных эффектов инсулина введение экзогенного инсулина может принести потенциальную пользу пациентам с болезнью Паркинсона.
Инсулин следует вводить интраназально, чтобы избежать воздействия на периферические уровни глюкозы. В экспериментах на моделях грызунов установлено, что интраназальный инсулин улучшал активацию и восстановление нейрональных стволовых клеток, разрастание дендритов и оказывал нейропротективное воздействие в условиях окислительного стресса [78].
В двойном слепом плацебо-контролируемом пилотном клиническом исследовании у пациентов с болезнью Альцгеймера продемонстрировано, что интраназальное введение инсулина улучшало память и сохраняло познавательные способности [79].
Однако следует отметить, что постоянное использование препаратов инсулина может способствовать десенсибилизации инсулина в головном мозге [18].
Агонисты рецепторов глюкагоноподобного пептида 1
К наиболее перспективным методам лечения следует отнести агонисты рецепторов глюкагоноподобного пептида 1 (ГПП-1). Данные препараты не влияют на рецепторы инсулина и таким образом предотвращают снижение чувствительности к инсулину с течением времени [80, 81]. Кроме того, они способны повторно сенсибилизировать передачу сигналов инсулина [82], не влияют на уровень глюкозы в крови при нормогликемии [83]. Именно поэтому можно не опасаться назначать их пациентам c болезнью Паркинсона в отсутствие сахарного диабета 2 типа [11].
В центральной нервной системе рецепторы ГПП-1 в основном экспрессируются на поверхности пирамидных нейронов коры или гиппокампа и нейронах Пуркинье в мозжечке и черной субстанции [84].
Cинтетические агонисты рецепторов ГПП-1 характеризуются более длительным периодом полувыведения по сравнению с эндогенным ГПП-1 [85]. Эти препараты связываются с рецепторами ГПП-1 и активируют путь PI3K/Akt, что приводит к инактивации микроглиальных клеток, снижению концентрации медиаторов воспаления, агрегации α-синуклеина [86], апоптоза [87] (cм. рис. 2).
Через путь MAPK агонисты рецепторов ГПП-1 также активируют экспрессию генов, участвующих в росте и репарации клеток, улучшая нейрозащиту от стрессовых факторов, таких как α-синуклеин и медиаторы воспаления [78].
В исследованиях агониста рецепторов ГПП-1 эксенатида получены многообещающие результаты в отношении замедления прогрессирования болезни Паркинсона, индуцированной у мышей [88].
В ходе открытого клинического испытания зафиксировано значимое улучшение моторных и когнитивных проявлений болезни Паркинсона [89].
При этом положительные эффекты терапии эксенатидом сохранялись через год после его отмены [90].
В двойном слепом плацебо-контролируемом исследовании подтвердилась концепция, согласно которой миметики ГПП-1 могут нормализовать передачу сигналов инсулина в головном мозге и изменять прогрессирование заболевания.
Проводятся также клинические испытания у пациентов с болезнью Паркинсона таких препаратов, как ликсисенатид [18], лираглутид [11] и семаглутид [91].
Тиазолидиндионы
Тиазолидиндионы – еще один класс пероральных сахароснижающих средств, продемонстрировавший значительное снижение риска развития болезни Паркинсона у страдающих сахарным диабетом 2 типа [92, 93]. В первую очередь они воздействуют на рецептор γ-активатора пролифератора пероксисом, модулируя транскрипцию генов, отвечающих за чувствительность к инсулину [7, 94].
Кроме того, считается, что тиазолидиндионы подавляют активацию микроглии и уменьшают окислительный стресс в нейронах, улучшая функцию митохондрий [95] и предотвращая нейродегенерацию.
Однако в недавнем двойном слепом клиническом исследовании фазы II было отмечено, что применение пиоглитазона в течение года значительно не влияло на снижение риска прогрессирования болезни Паркинсона по сравнению с использованием плацебо [96].
Эффективность тиазолидиндионов как потенциального варианта лечения болезни Паркинсона может быть ограничена из-за возможности развития побочных эффектов со стороны сердечно-сосудистой, костной (переломы) и мочевыделительной систем (рак мочевого пузыря) [6].
Метформин
Метформин помимо сахароснижающего обладает плейотропным действием и потенциально замедляет старение, воздействуя на митохондриальный метаболизм и передачу сигналов инсулина [97]. В исследованиях последних лет доказана способность метформина быстро проникать через гематоэнцефалический барьер [98] и обеспечивать нейрозащиту от инсульта, когнитивных нарушений, болезни Хантингтона, а также потенциально предотвращать деменцию [99]. Метформин может снижать фосфорилирование и агрегацию α-синуклеина, влиять на клеточные процессы, связанные с возрастными состояниями, включая воспаление и аутофагию. Кроме того, он способен восстанавливать физиологические молекулярные функции, нарушенные генетическими мутациями, ассоциированными с болезнью Паркинсона (Parkin, PINK1, DJ1, SNCA и LRRK2), и влиять на познавательные процессы.
В недавнем исследовании с участием пожилых ветеранов США, страдающих сахарным диабетом 2 типа, оценивалась связь между продолжительностью воздействия метформина и риском развития болезни Паркинсона. В ходе исследования установлено, что снижение риска возникновения этого нейродегенеративного заболевания могло быть обусловлено длительной терапией метформином [99].
Известно также, что длительный прием метформина приводил к дефициту витамина B12 и, как следствие, ухудшению состояния больных [100–102].
Агонисты рецепторов глюкозозависимого инсулинотропного полипептида и двойные агонисты
Некоторые методы лечения воздействуют на другие медиаторы инсулинового пути, в частности глюкозозависимый инсулинотропный полипептид (ГИП). ГИП – это инкретин, гормон, который участвует в снижении уровня глюкозы в крови и запускает различные нисходящие пути инсулина, способствуя его биосинтезу и секреции [80]. При тестировании на мышиной модели болезни Паркинсона аналог ГИП уменьшал хронический воспалительный ответ в головном мозге и окислительный стресс, оказывал нейропротективное воздействие на дофаминергические нейроны черной субстанции [11].
Определенные надежды связывают с разработкой двойных агонистов – и рецепторов ГИП, и рецепторов ГПП-1, которые обладают сходным сродством в отношении активации рецепторов как ГИП, так и ГПП-1, способных преодолевать гематоэнцефалический барьер и оказывать нейропротективное воздействие [103]. По сравнению с агонистом рецепторов ГПП-1 двойной агонист более эффективен в отношении стимуляции передачи сигналов инсулина [104] и обладает меньшим количеством побочных эффектов [105].
Двойной агонист рецепторов ГПП-1/ГИП может снижать активацию микроглии, что способствует выработке дофамина и обеспечивает нейрозащиту дофаминергических нейронов [106]. Кроме того, сообщалось о снижении нейровоспаления и восстановлении чувствительности к инсулину у грызунов, получавших 1-метил-4-фенил-1,2,3,6-тетрагидропиридин, после применения агониста рецепторов ГПП-1/ГИП [107].
Субетта
В качестве перспективного направления коррекции инсулинорезистентного состояния рассматривается оригинальный препарат на основе технологически обработанных антител Субетта.
Препарат активирует рецептор инсулина, увеличивает инсулинзависимый метаболизм глюкозы, повышая захват глюкозы миоцитами на 43% [108]. Второй компонент препарата позволяет корректировать эндотелиальную дисфункцию за счет стимулирования синтеза оксида азота (NO). Способность технологически обработанных антител к эндотелиальной NO-синтазе нивелировать дефицит NO приводит к снижению реактивности сосудов, уменьшению сосудистого спазма, нормализации уровня артериального давления и улучшению периферической микроциркуляции [109, 110]. Сосудистые эффекты препарата позволяют профилактировать развитие осложнений сахарного диабета.
В исследовании эффективности и безопасности препарата Субетта у пациентов с предиабетом было продемонстрировано значимое снижение уровня двухчасовой глюкозы плазмы до нормальных значений, что свидетельствовало о возможностях профилактики прогрессирования нарушений углеводного обмена [111].
Заключение
Эпидемиологические и экспериментальные данные подтверждают связь между болезнью Паркинсона и сахарным диабетом 2 типа. Патофизиология нейродегенерации при болезни Паркинсона гетерогенна и сложна.
Сахарный диабет 2 типа увеличивает риск возникновения болезни Паркинсона, коррелирует с быстрым прогрессированием заболевания, тяжестью моторного и когнитивного дефицита.
В основе патогенеза как сахарного диабета 2 типа, так и болезни Паркинсона лежат аберрантное накопление белка, лизосомная и митохондриальная дисфункция, хроническое системное воспаление.
Ключевую роль в патогенезе болезни Паркинсона играет инсулинорезистентность.
Нарушение передачи сигналов инсулина в клетках головного мозга потенциально может предрасполагать к возникновению и прогрессированию нейродегенерации.
Общие патофизиологические процессы рассматриваемых нозологий обусловливают возможность применения сахароснижающих препаратов в качестве нейропротективных средств. Таким образом, может быть разработана новая стратегия таргетной терапии, модифицирующей течение болезни Паркинсона.
A.Yu. Troshneva, O.S. Levin, MD, PhD, Prof., A.S. Ametov, MD, PhD, Prof.
Russian Medical Academy of Continuous Professional Education
Contact person: Anna Yu. Troshneva, troshneva@yandex.ru
In recent years, an emerging body of evidence has forged links between Parkinson's disease and type 2 diabetes mellitus.
Type 2 diabetes mellitus is associated with an increased risk for developing and faster progression of Parkinson's disease, correlates with the severity of motor and cognitive deficits in this group of patients.
One of the key links in pathogenesis of type 2 diabetes mellitus is insulin resistance. There is growing evidence that a process similar to peripheral insulin resistance occurs in the brain tissue of patients with Parkinson's disease.
Today, there are no available effective disease modifying treatments for Parkinson's disease. The use of antidiabetic drugs as neuroprotective agents opens up the possibility of developing a new strategy for targeted therapy that modifies the course of Parkinson's disease.
This review examines the epidemiological, clinical, and pathophysiological aspects of the relationship between insulin resistance and Parkinson's disease, as well as potential ways of repurposing antidiabetic therapy for the treatment of Parkinson's disease.
Уважаемый посетитель uMEDp!
Уведомляем Вас о том, что здесь содержится информация, предназначенная исключительно для специалистов здравоохранения.
Если Вы не являетесь специалистом здравоохранения, администрация не несет ответственности за возможные отрицательные последствия, возникшие в результате самостоятельного использования Вами информации с портала без предварительной консультации с врачом.
Нажимая на кнопку «Войти», Вы подтверждаете, что являетесь врачом или студентом медицинского вуза.