–Р–љ–Њ–Љ–∞–ї–Є—П –≠–±—И—В–µ–є–љ–∞
- –Р–љ–љ–Њ—В–∞—Ж–Є—П
- –°—В–∞—В—М—П
- –°—Б—Л–ї–Ї–Є
- English
–Т –Ї–∞—З–µ—Б—В–≤–µ –≤–Њ–Ј–Љ–Њ–ґ–љ—Л—Е –њ—А–Є—З–Є–љ —А–∞–Ј–≤–Є—В–Є—П –њ–Њ—А–Њ–Ї–∞ –љ–∞–Ј—Л–≤–∞—О—В –≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є–µ –Є —Б—А–µ–і–Њ–≤—Л–µ —Д–∞–Ї—В–Њ—А—Л (–≤–Є—А—Г—Б–љ—Л–µ –Є–љ—Д–µ–Ї—Ж–Є–Є, –њ—А–Є–Љ–µ–љ–µ–љ–Є–µ –±–µ–љ–Ј–Њ–і–Є–∞–Ј–µ–њ–Є–љ–Њ–≤, –њ—А–µ–њ–∞—А–∞—В–Њ–≤ –ї–Є—В–Є—П). –Ъ–ї–Є–љ–Є—З–µ—Б–Ї–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ –њ—А–Њ—П–≤–ї—П–µ—В—Б—П —Ж–Є–∞–љ–Њ–Ј–Њ–Љ, –Њ–і—Л—И–Ї–Њ–є, —Б–µ—А–і–µ—З–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М—О, –љ–∞—А—Г—И–µ–љ–Є–µ–Љ —А–Є—В–Љ–∞ —Б–µ—А–і—Ж–∞. –Ь–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є –Њ—В–Љ–µ—З–∞—О—В—Б—П –∞–љ–Њ–Љ–∞–ї–Є–Є —В—А–Є–Ї—Г—Б–њ–Є–і–∞–ї—М–љ–Њ–≥–Њ –Ї–ї–∞–њ–∞–љ–∞, —Б–Њ—Б–Њ—З–Ї–Њ–≤—Л—Е –Љ—Л—И—Ж –Є —Е–Њ—А–і–∞–ї—М–љ–Њ–≥–Њ –∞–њ–њ–∞—А–∞—В–∞, –≥–Є–њ–µ—А—В—А–Њ—Д–Є—П –Љ–Є–Њ–Ї–∞—А–і–∞ –њ—А–∞–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞, –Є—Б—В–Њ–љ—З–µ–љ–Є–µ —Б—В–µ–љ–Њ–Ї –њ—А–∞–≤–Њ–≥–Њ –њ—А–µ–і—Б–µ—А–і–Є—П –Є –∞—В—А–Є–∞–ї–Є–Ј–Њ–≤–∞–љ–љ–Њ–є —З–∞—Б—В–Є –њ—А–∞–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞. –†–µ—И–∞—О—Й–Є–Љ –і–ї—П –і–Є–∞–≥–љ–Њ–Ј–∞ —П–≤–ї—П–µ—В—Б—П —Г–ї—М—В—А–∞–Ј–≤—Г–Ї–Њ–≤–Њ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ, –њ–Њ –і–∞–љ–љ—Л–Љ –Ї–Њ—В–Њ—А–Њ–≥–Њ –Њ–±–љ–∞—А—Г–ґ–Є–≤–∞—О—В —Б–Љ–µ—Й–µ–љ–Є–µ –њ—А–∞–≤–Њ–≥–Њ –∞—В—А–Є–Њ–≤–µ–љ—В—А–Є–Ї—Г–ї—П—А–љ–Њ–≥–Њ –Ї–Њ–ї—М—Ж–∞. –°—В–µ–њ–µ–љ—М —Б–Љ–µ—Й–µ–љ–Є—П –Њ–њ—А–µ–і–µ–ї—П–µ—В —В—П–ґ–µ—Б—В—М –њ–Њ—А–Њ–Ї–∞. –Р–Љ–њ–ї–Є—В—Г–і–∞ –і–≤–Є–ґ–µ–љ–Є—П —В—А–Є–Ї—Г—Б–њ–Є–і–∞–ї—М–љ–Њ–≥–Њ –Ї–ї–∞–њ–∞–љ–∞ —Г–≤–µ–ї–Є—З–µ–љ–∞. –Ч–∞–Ї—А—Л—В–Є–µ —Н—В–Њ–≥–Њ –Ї–ї–∞–њ–∞–љ–∞ –≤ –Њ—В–ї–Є—З–Є–µ –Њ—В –Љ–Є—В—А–∞–ї—М–љ–Њ–≥–Њ –њ—А–Њ–Є—Б—Е–Њ–і–Є—В –њ–Њ–Ј–і–љ–µ–µ. –†–∞–Ј–љ–Є—Ж–∞ 40 –Љ—Б –Љ–µ–ґ–і—Г —В–Њ—З–Ї–∞–Љ–Є –Ј–∞–Ї—А—Л—В–Є—П —В—А–Є–Ї—Г—Б–њ–Є–і–∞–ї—М–љ–Њ–≥–Њ –Є –Љ–Є—В—А–∞–ї—М–љ–Њ–≥–Њ –Ї–ї–∞–њ–∞–љ–Њ–≤ вАУ –і–Є–∞–≥–љ–Њ—Б—В–Є—З–µ—Б–Ї–Є –≤–∞–ґ–љ—Л–є –њ—А–Є–Ј–љ–∞–Ї –∞–љ–Њ–Љ–∞–ї–Є–Є –≠–±—И—В–µ–є–љ–∞. –Ґ–∞–Ї—В–Є–Ї–∞ –≤—А–∞—З–∞ –Њ–њ—А–µ–і–µ–ї—П–µ—В—Б—П –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–є –Ї–∞—А—В–Є–љ–Њ–є.
–Т –Ї–∞—З–µ—Б—В–≤–µ –≤–Њ–Ј–Љ–Њ–ґ–љ—Л—Е –њ—А–Є—З–Є–љ —А–∞–Ј–≤–Є—В–Є—П –њ–Њ—А–Њ–Ї–∞ –љ–∞–Ј—Л–≤–∞—О—В –≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є–µ –Є —Б—А–µ–і–Њ–≤—Л–µ —Д–∞–Ї—В–Њ—А—Л (–≤–Є—А—Г—Б–љ—Л–µ –Є–љ—Д–µ–Ї—Ж–Є–Є, –њ—А–Є–Љ–µ–љ–µ–љ–Є–µ –±–µ–љ–Ј–Њ–і–Є–∞–Ј–µ–њ–Є–љ–Њ–≤, –њ—А–µ–њ–∞—А–∞—В–Њ–≤ –ї–Є—В–Є—П). –Ъ–ї–Є–љ–Є—З–µ—Б–Ї–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ –њ—А–Њ—П–≤–ї—П–µ—В—Б—П —Ж–Є–∞–љ–Њ–Ј–Њ–Љ, –Њ–і—Л—И–Ї–Њ–є, —Б–µ—А–і–µ—З–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М—О, –љ–∞—А—Г—И–µ–љ–Є–µ–Љ —А–Є—В–Љ–∞ —Б–µ—А–і—Ж–∞. –Ь–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є –Њ—В–Љ–µ—З–∞—О—В—Б—П –∞–љ–Њ–Љ–∞–ї–Є–Є —В—А–Є–Ї—Г—Б–њ–Є–і–∞–ї—М–љ–Њ–≥–Њ –Ї–ї–∞–њ–∞–љ–∞, —Б–Њ—Б–Њ—З–Ї–Њ–≤—Л—Е –Љ—Л—И—Ж –Є —Е–Њ—А–і–∞–ї—М–љ–Њ–≥–Њ –∞–њ–њ–∞—А–∞—В–∞, –≥–Є–њ–µ—А—В—А–Њ—Д–Є—П –Љ–Є–Њ–Ї–∞—А–і–∞ –њ—А–∞–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞, –Є—Б—В–Њ–љ—З–µ–љ–Є–µ —Б—В–µ–љ–Њ–Ї –њ—А–∞–≤–Њ–≥–Њ –њ—А–µ–і—Б–µ—А–і–Є—П –Є –∞—В—А–Є–∞–ї–Є–Ј–Њ–≤–∞–љ–љ–Њ–є —З–∞—Б—В–Є –њ—А–∞–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞. –†–µ—И–∞—О—Й–Є–Љ –і–ї—П –і–Є–∞–≥–љ–Њ–Ј–∞ —П–≤–ї—П–µ—В—Б—П —Г–ї—М—В—А–∞–Ј–≤—Г–Ї–Њ–≤–Њ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ, –њ–Њ –і–∞–љ–љ—Л–Љ –Ї–Њ—В–Њ—А–Њ–≥–Њ –Њ–±–љ–∞—А—Г–ґ–Є–≤–∞—О—В —Б–Љ–µ—Й–µ–љ–Є–µ –њ—А–∞–≤–Њ–≥–Њ –∞—В—А–Є–Њ–≤–µ–љ—В—А–Є–Ї—Г–ї—П—А–љ–Њ–≥–Њ –Ї–Њ–ї—М—Ж–∞. –°—В–µ–њ–µ–љ—М —Б–Љ–µ—Й–µ–љ–Є—П –Њ–њ—А–µ–і–µ–ї—П–µ—В —В—П–ґ–µ—Б—В—М –њ–Њ—А–Њ–Ї–∞. –Р–Љ–њ–ї–Є—В—Г–і–∞ –і–≤–Є–ґ–µ–љ–Є—П —В—А–Є–Ї—Г—Б–њ–Є–і–∞–ї—М–љ–Њ–≥–Њ –Ї–ї–∞–њ–∞–љ–∞ —Г–≤–µ–ї–Є—З–µ–љ–∞. –Ч–∞–Ї—А—Л—В–Є–µ —Н—В–Њ–≥–Њ –Ї–ї–∞–њ–∞–љ–∞ –≤ –Њ—В–ї–Є—З–Є–µ –Њ—В –Љ–Є—В—А–∞–ї—М–љ–Њ–≥–Њ –њ—А–Њ–Є—Б—Е–Њ–і–Є—В –њ–Њ–Ј–і–љ–µ–µ. –†–∞–Ј–љ–Є—Ж–∞ 40 –Љ—Б –Љ–µ–ґ–і—Г —В–Њ—З–Ї–∞–Љ–Є –Ј–∞–Ї—А—Л—В–Є—П —В—А–Є–Ї—Г—Б–њ–Є–і–∞–ї—М–љ–Њ–≥–Њ –Є –Љ–Є—В—А–∞–ї—М–љ–Њ–≥–Њ –Ї–ї–∞–њ–∞–љ–Њ–≤ вАУ –і–Є–∞–≥–љ–Њ—Б—В–Є—З–µ—Б–Ї–Є –≤–∞–ґ–љ—Л–є –њ—А–Є–Ј–љ–∞–Ї –∞–љ–Њ–Љ–∞–ї–Є–Є –≠–±—И—В–µ–є–љ–∞. –Ґ–∞–Ї—В–Є–Ї–∞ –≤—А–∞—З–∞ –Њ–њ—А–µ–і–µ–ї—П–µ—В—Б—П –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–є –Ї–∞—А—В–Є–љ–Њ–є.
–Ю–њ—А–µ–і–µ–ї–µ–љ–Є–µ
–Р–љ–Њ–Љ–∞–ї–Є—П –≠–±—И—В–µ–є–љ–∞ (Q 22.5)¬†вАУ —А–µ–і–Ї–Є–є –≤—А–Њ–ґ–і–µ–љ–љ—Л–є –њ–Њ—А–Њ–Ї, –Ј–∞–Ї–ї—О—З–∞—О—Й–Є–є—Б—П –≤¬†–∞–њ–Є–Ї–∞–ї—М–љ–Њ–Љ —Б–Љ–µ—Й–µ–љ–Є–Є –њ—А–∞–≤–Њ–≥–Њ –∞—В—А–Є–Њ–≤–µ–љ—В—А–Є–Ї—Г–ї—П—А–љ–Њ–≥–Њ –Ї–Њ–ї—М—Ж–∞ —Б¬†–∞–љ–Њ–Љ–∞–ї–Є–µ–є —А–∞–Ј–≤–Є—В–Є—П —В—А–Є–Ї—Г—Б–њ–Є–і–∞–ї—М–љ–Њ–≥–Њ –Ї–ї–∞–њ–∞–љ–∞ –Є¬†–љ–µ—А–µ–і–Ї–Њ —Б–Њ—Б—Г—Й–µ—Б—В–≤–Њ–≤–∞–љ–Є–µ–Љ –і—А—Г–≥–Є—Е –њ–Њ—А–Њ–Ї–Њ–≤ —Б–µ—А–і—Ж–∞ [1]. –Я–Њ—А–Њ–Ї –≤–њ–µ—А–≤—Л–µ –±—Л–ї –Њ–њ–Є—Б–∞–љ –Т–Є–ї—М–≥–µ–ї—М–Љ–Њ–Љ –≠–±—И—В–µ–є–љ–Њ–Љ –≤¬†1866¬†–≥.¬†—Г 19-–ї–µ—В–љ–µ–≥–Њ —О–љ–Њ—И–Є —Б¬†–Њ–і—Л—И–Ї–Њ–є, —В–∞—Е–Є–Ї–∞—А–і–Є–µ–є, —Ж–Є–∞–љ–Њ–Ј–Њ–Љ, —А–∞—Б—И–Є—А–µ–љ–Є–µ–Љ —П—А–µ–Љ–љ—Л—Е –≤–µ–љ –Є¬†–≥—А–∞–љ–Є—Ж —Б–µ—А–і—Ж–∞.
–Р–љ–Њ–Љ–∞–ї–Є—П –≠–±—И—В–µ–є–љ–∞ —З–∞—Б—В–Њ —Б–Њ—З–µ—В–∞–µ—В—Б—П —Б¬†–і–µ—Д–µ–Ї—В–Њ–Љ –Љ–µ–ґ–њ—А–µ–і—Б–µ—А–і–љ–Њ–є –њ–µ—А–µ–≥–Њ—А–Њ–і–Ї–Є (90% –≤—Б–µ—Е —Б–ї—Г—З–∞–µ–≤), —Б—В–µ–љ–Њ–Ј–Њ–Љ –Є–ї–Є –∞—В—А–µ–Ј–Є–µ–є –ї–µ–≥–Њ—З–љ–Њ–є –∞—А—В–µ—А–Є–Є (20вАУ25%), —Б–Є–љ–і—А–Њ–Љ–Њ–Љ –Т–Њ–ї—М—Д—Д–∞¬†вАУ –Я–∞—А–Ї–Є–љ—Б–Њ–љ–∞¬†вАУ –£–∞–є—В–∞ (20%). –Я—А–Є –ї–µ–≤–Њ-—В—А–∞–љ—Б–њ–Њ–Ј–Є—Ж–Є–Є –Љ–∞–≥–Є—Б—В—А–∞–ї—М–љ—Л—Е —Б–Њ—Б—Г–і–Њ–≤ (L-TGA) —В—А–Є–Ї—Г—Б–њ–Є–і–∞–ї—М–љ—Л–є –Ї–ї–∞–њ–∞–љ —А–∞—Б–њ–Њ–ї–Њ–ґ–µ–љ —Б–ї–µ–≤–∞, —З—В–Њ¬†–≥–Њ–≤–Њ—А–Є—В –Њ¬†—Н–±—И—В–µ–љ–Њ–Є–і–љ–Њ–є –∞–љ–Њ–Љ–∞–ї–Є–Є [2].
–†–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–љ–Њ—Б—В—М –Є¬†–≤–Њ–Ј–Љ–Њ–ґ–љ—Л–µ –њ—А–Є—З–Є–љ–љ—Л–µ —Д–∞–Ї—В–Њ—А—Л
–Р–љ–Њ–Љ–∞–ї–Є—П –≠–±—И—В–µ–є–љ–∞ —А–µ–≥–Є—Б—В—А–Є—А—Г–µ—В—Б—П —Б¬†—З–∞—Б—В–Њ—В–Њ–є 1:200 000 –ґ–Є–≤–Њ—А–Њ–ґ–і–µ–љ–љ—Л—Е, —Б–Њ—Б—В–∞–≤–ї—П—П 0,3вАУ0,6% –≤—Б–µ—Е —Б–ї—Г—З–∞–µ–≤ –≤—А–Њ–ґ–і–µ–љ–љ—Л—Е –њ–Њ—А–Њ–Ї–Њ–≤ —Б–µ—А–і—Ж–∞. –Ь—Л –љ–∞–±–ї—О–і–∞–ї–Є —И–µ—Б—В—М –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–∞–љ–Њ–Љ–∞–ї–Є–µ–є –≠–±—И—В–µ–є–љ–∞. –Я–Њ¬†–љ–∞—И–Є–Љ –і–∞–љ–љ—Л–Љ, –Ј–∞ 42¬†–≥–Њ–і–∞ —А–∞–±–Њ—В—Л –њ–∞—В–Њ–ї–Њ–≥–Њ–∞–љ–∞—В–Њ–Љ–Є—З–µ—Б–Ї–Њ–≥–Њ –Њ—В–і–µ–ї–µ–љ–Є—П –Њ–±–ї–∞—Б—В–љ–Њ–є –±–Њ–ї—М–љ–Є—Ж—Л —Н—В–Њ—В –њ–Њ—А–Њ–Ї –і–Є–∞–≥–љ–Њ—Б—В–Є—А–Њ–≤–∞–љ –≤¬†–і–≤—Г—Е —Б–ї—Г—З–∞—П—Е (–±–Њ–ї–µ–µ —З–µ–Љ –љ–∞¬†6000 —Б–µ–Ї—Ж–Є–Њ–љ–љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є)¬†вАУ —Г¬†–≤–Ј—А–Њ—Б–ї–Њ–≥–Њ –Є¬†—А–µ–±–µ–љ–Ї–∞. –Ч–∞ –њ–µ—А–Є–Њ–і 1979вАУ2019¬†–≥–≥. –≤¬†–Ї–∞–±–Є–љ–µ—В–∞—Е —Г–ї—М—В—А–∞–Ј–≤—Г–Ї–Њ–≤–Њ–є –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–Є –Љ–љ–Њ–≥–Њ–њ—А–Њ—Д–Є–ї—М–љ—Л—Е –і–µ—В—Б–Ї–Є—Е –±–Њ–ї—М–љ–Є—Ж –∞–љ–Њ–Љ–∞–ї–Є—П –≠–±—И—В–µ–є–љ–∞ –≤—Л—П–≤–ї–µ–љ–∞ –љ–∞–Љ–Є –≤¬†–і–≤—Г—Е —Б–ї—Г—З–∞—П—Е –љ–∞¬†–±–Њ–ї–µ–µ —З–µ–Љ 32 000 —Н—Е–Њ–Ї–∞—А–і–Є–Њ–≥—А–∞—Д–Є–є. –Т¬†–≥–Њ—А–Њ–і—Б–Ї–Њ–є –і–µ—В—Б–Ї–Њ–є –њ–Њ–ї–Є–Ї–ї–Є–љ–Є–Ї–µ –Ј–∞—Д–Є–Ї—Б–Є—А–Њ–≤–∞–љ–Њ –µ—Й–µ –і–≤–∞ —Б–ї—Г—З–∞—П –љ–∞¬†24¬†000¬†—Н—Е–Њ–Ї–∞—А–і–Є–Њ–≥—А–∞—Д–Є–є. –£¬†–љ–∞—И–Є—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –∞–љ–Њ–Љ–∞–ї–Є—П –≠–±—И—В–µ–є–љ–∞ —Б–Њ—З–µ—В–∞–ї–∞—Б—М —Б¬†–і–µ—Д–µ–Ї—В–Њ–Љ –Љ–µ–ґ–њ—А–µ–і—Б–µ—А–і–љ–Њ–є –њ–µ—А–µ–≥–Њ—А–Њ–і–Ї–Є (4/6) –Є¬†—Б–Є–љ–і—А–Њ–Љ–Њ–Љ –Т–Њ–ї—М—Д—Д–∞¬†вАУ –Я–∞—А–Ї–Є–љ—Б–Њ–љ–∞¬†вАУ –£–∞–є—В–∞ (1/6). –Т–Њ –≤—Б–µ—Е –љ–∞—И–Є—Е –љ–∞–±–ї—О–і–µ–љ–Є—П—Е –Є–Љ–µ–ї–Є –Љ–µ—Б—В–Њ —Б–њ–Њ—А–∞–і–Є—З–µ—Б–Ї–Є–µ –≤–∞—А–Є–∞–љ—В—Л. –Т¬†–ї–Є—В–µ—А–∞—В—Г—А–µ –Њ–њ–Є—Б–∞–љ—Л —Б–µ–Љ–µ–є–љ—Л–µ –≤–∞—А–Є–∞–љ—В—Л, —Е–Њ—В—П –њ–Њ–і—З–µ—А–Ї–Є–≤–∞–µ—В—Б—П —Б–њ–Њ—А–∞–і–Є—З–љ–Њ—Б—В—М –њ–Њ–і–∞–≤–ї—П—О—Й–µ–≥–Њ –±–Њ–ї—М—И–Є–љ—Б—В–≤–∞ —Б–ї—Г—З–∞–µ–≤. –Т—Л—Б–Њ–Ї–∞—П —З–∞—Б—В–Њ—В–∞ –∞–љ–Њ–Љ–∞–ї–Є–Є –≠–±—И—В–µ–є–љ–∞ (6:100) –Ј–∞—А–µ–≥–Є—Б—В—А–Є—А–Њ–≤–∞–љ–∞ —Г¬†–і–µ—В–µ–є, —А–Њ–ґ–і–µ–љ–љ—Л—Е –Љ–∞—В–µ—А—П–Љ–Є —Б¬†—В–µ–Љ –ґ–µ –њ–Њ—А–Њ–Ї–Њ–Љ. –Х—Б–ї–Є –љ–Њ—Б–Є—В–µ–ї–µ–Љ –∞–љ–Њ–Љ–∞–ї–Є–Є —П–≤–ї—П–µ—В—Б—П –Њ—В–µ—Ж, –≤–µ—А–Њ—П—В–љ–Њ—Б—В—М —А–Њ–ґ–і–µ–љ–Є—П —А–µ–±–µ–љ–Ї–∞ —Б¬†–∞–љ–Њ–Љ–∞–ї–Є–µ–є –≠–±—И—В–µ–є–љ–∞ —Б–љ–Є–ґ–∞–µ—В—Б—П –≤¬†–і–µ—Б—П—В—М —А–∞–Ј (6:1000). –Ґ–µ–Љ –љ–µ¬†–Љ–µ–љ–µ–µ –Њ–љ–∞ –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ –≤—Л—И–µ, —З–µ–Љ –≤¬†–Њ–±—Й–µ–є –њ–Њ–њ—Г–ї—П—Ж–Є–Є.
–°–њ–µ—Ж–Є—Д–Є—З–µ—Б–Ї–Њ–≥–Њ¬†–≥–µ–љ–∞, –Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ–≥–Њ –Ј–∞ —А–∞–Ј–≤–Є—В–Є–µ –∞–љ–Њ–Љ–∞–ї–Є–Є –≠–±—И—В–µ–є–љ–∞, –љ–µ¬†–≤—Л—П–≤–ї–µ–љ–Њ. –У–µ–љ–∞–Љ–Є-–Ї–∞–љ–і–Є–і–∞—В–∞–Љ–Є —П–≤–ї—П—О—В—Б—П MYH7 –Є¬†NKX2-5. –£¬†–љ–Њ—Б–Є—В–µ–ї–µ–є –њ–Њ—А–Њ–Ї–∞ –љ–µ—А–µ–і–Ї–Њ –≤—Л—П–≤–ї—П–µ—В—Б—П –Љ–Є—Б—Б–µ–љ—Б-–Љ—Г—В–∞—Ж–Є—П FLNA (—Д–Є–ї–∞–Љ–Є–љ –Р, –∞–Ї—В–Є–љ-—Б–≤—П–Ј—Л–≤–∞—О—Й–Є–є –њ—А–Њ—В–µ–Є–љ) –љ–∞¬†Xq28 [3вАУ6]. –£¬†—А–Њ–і—Б—В–≤–µ–љ–љ–Є–Ї–Њ–≤ —А–µ–і–Ї–Њ –Њ–±–љ–∞—А—Г–ґ–Є–≤–∞—О—В—Б—П –Љ—Г—В–∞—Ж–Є–Є –Ї–∞—А–і–Є–∞–ї—М–љ–Њ–≥–Њ —Д–∞–Ї—В–Њ—А–∞ —В—А–∞–љ—Б–Ї—А–Є–њ—Ж–Є–Є NKX2-5, –і–µ–ї–µ—Ж–Є–Є 10p13-p14 –Є¬†1p34.3-p36.11 [7]. –Т¬†–Ї–∞—З–µ—Б—В–≤–µ –≤–Њ–Ј–Љ–Њ–ґ–љ—Л—Е –≤–љ–µ—И–љ–Є—Е —Д–∞–Ї—В–Њ—А–Њ–≤ –љ–∞–Ј—Л–≤–∞—О—В –≤–Є—А—Г—Б–љ—Л–µ –Є–љ—Д–µ–Ї—Ж–Є–Є, –њ—А–Є–Љ–µ–љ–µ–љ–Є–µ –±–µ—А–µ–Љ–µ–љ–љ—Л–Љ–Є –±–µ–љ–Ј–Њ–і–Є–∞–Ј–µ–њ–Є–љ–Њ–≤ –Є¬†–њ—А–µ–њ–∞—А–∞—В–Њ–≤ –ї–Є—В–Є—П [8]. –Я–Њ—Б–ї–µ–і–љ–Є–є —Б—З–Є—В–∞—О—В –Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ—Л–Љ –Љ–µ–љ–µ–µ —З–µ–Љ –Ј–∞ 2% —Б–ї—Г—З–∞–µ–≤ –∞–љ–Њ–Љ–∞–ї–Є–Є –≠–±—И—В–µ–є–љ–∞¬†[9].
–Я–∞—В–Њ–Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—П –Є¬†–љ–∞—А—Г—И–µ–љ–Є—П¬†–≥–µ–Љ–Њ–і–Є–љ–∞–Љ–Є–Ї–Є
–Т¬†–Ј–і–Њ—А–Њ–≤–Њ–Љ —Б–µ—А–і—Ж–µ –њ—А–∞–≤–Њ–µ –Є¬†–ї–µ–≤–Њ–µ –∞—В—А–Є–Њ–≤–µ–љ—В—А–Є–Ї—Г–ї—П—А–љ—Л–µ –Ї–Њ–ї—М—Ж–∞ —А–∞—Б–њ–Њ–ї–Њ–ґ–µ–љ—Л –љ–∞¬†–Њ–і–љ–Њ–Љ —Г—А–Њ–≤–љ–µ. –Ґ—А–µ—Е—Б—В–≤–Њ—А—З–∞—В—Л–є –Ї–ї–∞–њ–∞–љ —Б–Њ—Б—В–Њ–Є—В –Є–Ј¬†—В—А–µ—Е –ї–µ–њ–µ—Б—В–Ї–Њ–≤ (–њ–µ—А–µ–і–љ—П—П, –Ј–∞–і–љ—П—П –Є¬†—Б–µ–њ—В–∞–ї—М–љ–∞—П —Б—В–≤–Њ—А–Ї–Є). –Р–љ–Њ–Љ–∞–ї–Є—П –≠–±—И—В–µ–є–љ–∞¬†вАУ –љ–∞—А—Г—И–µ–љ–Є–µ —А–∞–Ј–≤–Є—В–Є—П —В—А–µ—Е—Б—В–≤–Њ—А—З–∞—В–Њ–≥–Њ –Ї–ї–∞–њ–∞–љ–∞ –Є¬†–њ—А–∞–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞ [10]:
- –≤¬†–њ–µ—А–Є–Њ–і —Н–Љ–±—А–Є–Њ–љ–∞–ї—М–љ–Њ–≥–Њ —А–∞–Ј–≤–Є—В–Є—П –љ–∞—А—Г—И–∞–µ—В—Б—П –Њ—В—Б–ї–∞–Є–≤–∞–љ–Є–µ –Ј–∞—З–∞—В–Ї–∞ —Б—В–≤–Њ—А–Ї–Є –Ї–ї–∞–њ–∞–љ–∞ –Њ—В¬†–Љ–Є–Њ–Ї–∞—А–і–∞. –С√≥–ї—М—И–µ–є —Б–≤–Њ–µ–є —З–∞—Б—В—М—О –Њ–љ–∞ –Њ—Б—В–∞–µ—В—Б—П —Б–њ–∞—П–љ–љ–Њ–є —Б¬†–Љ–Є–Њ–Ї–∞—А–і–Њ–Љ;
- –њ—А–∞–≤–Њ–µ –∞—В—А–Є–Њ–≤–µ–љ—В—А–Є–Ї—Г–ї—П—А–љ–Њ–µ –Ї–Њ–ї—М—Ж–Њ —Б–Љ–µ—Й–∞–µ—В—Б—П –≤¬†—Б—В–Њ—А–Њ–љ—Г –≤–µ—А—Е—Г—И–Ї–Є —Б–µ—А–і—Ж–∞ (—Б–µ–њ—В–∞–ї—М–љ–∞—П —Б—В–≤–Њ—А–Ї–∞ > –Ј–∞–і–љ–µ–є > –њ–µ—А–µ–і–љ–µ–є);
- —З–∞—Б—В—М –њ—А–∞–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞, –Њ–Ї–∞–Ј–∞–≤—И–∞—П—Б—П –љ–∞–і –Ї–ї–∞–њ–∞–љ–љ—Л–Љ –Ї–Њ–ї—М—Ж–Њ–Љ, —А–∞—Б—И–Є—А—П–µ—В—Б—П, –њ—А–Є–љ–Є–Љ–∞–µ—В –љ–∞¬†—Б–µ–±—П —Д—Г–љ–Ї—Ж–Є—О –њ—А–µ–і—Б–µ—А–і–Є—П (–∞—В—А–Є–∞–ї–Є–Ј—Г–µ—В—Б—П), —Б—В–µ–љ–Ї–Є –∞—В—А–Є–∞–ї–Є–Ј–Њ–≤–∞–љ–љ–Њ–є —З–∞—Б—В–Є –Є—Б—В–Њ–љ—З–∞—О—В—Б—П;
- —А–∞—Б—И–Є—А—П–µ—В—Б—П –њ—А–∞–≤–Њ–µ –∞—В—А–Є–Њ–≤–µ–љ—В—А–Є–Ї—Г–ї—П—А–љ–Њ–µ –Ї–Њ–ї—М—Ж–Њ.
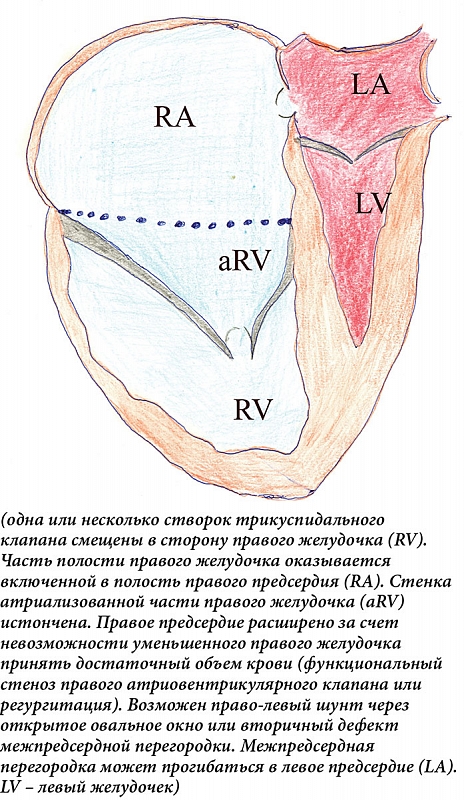
–Ґ–Њ—З–Ї–∞ –Љ–∞–Ї—Б–Є–Љ–∞–ї—М–љ–Њ–≥–Њ —Б–Љ–µ—Й–µ–љ–Є—П –љ–∞—Е–Њ–і–Є—В—Б—П –љ–∞¬†–Ї–Њ–Љ–Є—Б—Б—Г—А–µ –Љ–µ–ґ–і—Г –Ј–∞–і–љ–µ–є –Є¬†—Б–µ–њ—В–∞–ї—М–љ–Њ–є —Б—В–≤–Њ—А–Ї–∞–Љ–Є. –Я–µ—А–µ–і–љ—П—П —Б—В–≤–Њ—А–Ї–∞ —Г–≤–µ–ї–Є—З–µ–љ–∞ –≤¬†—А–∞–Ј–Љ–µ—А–∞—Е, –Љ–Њ–ґ–µ—В –±—Л—В—М –њ–µ—А—Д–Њ—А–Є—А–Њ–≤–∞–љ–∞, –і–µ—Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–∞. –Э–µ—Б–±–∞–ї–∞–љ—Б–Є—А–Њ–≤–∞–љ–љ–∞—П –і–µ—Д–Њ—А–Љ–∞—Ж–Є—П –њ—А–Є–≤–Њ–і–Є—В –Ї¬†–≤—А–∞—Й–∞—В–µ–ї—М–љ–Њ–Љ—Г —Б–Љ–µ—Й–µ–љ–Є—О —В—А–Є–Ї—Г—Б–њ–Є–і–∞–ї—М–љ–Њ–≥–Њ –Ї–ї–∞–њ–∞–љ–∞ –≤¬†–≤—Л–љ–Њ—Б–љ–Њ–є —В—А–∞–Ї—В –њ—А–∞–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞, –≤—Л–Ј—Л–≤–∞—П –Њ–±—Б—В—А—Г–Ї—Ж–Є—О. –•–Њ—А–і–∞–ї—М–љ—Л–µ –љ–Є—В–Є –њ–µ—А–µ–і–љ–µ–є —Б—В–≤–Њ—А–Ї–Є —Г–Ї–Њ—А–Њ—З–µ–љ—Л, —А–∞–Ј–≤–Є—В—Л –њ–ї–Њ—Е–Њ. –Я—А–∞–≤—Л–є –ґ–µ–ї—Г–і–Њ—З–µ–Ї —А–∞–Ј–і–µ–ї–µ–љ –љ–∞¬†–і–≤–µ —З–∞—Б—В–Є¬†вАУ –≤–Њ–≤–ї–µ—З–µ–љ–љ—Г—О –≤¬†–њ–Њ—А–Њ–Ї (–∞—В—А–Є–∞–ї–Є–Ј–Њ–≤–∞–љ–љ—Г—О) —З–∞—Б—В—М –њ—А–∞–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞ –Є¬†—Б–Њ–±—Б—В–≤–µ–љ–љ–Њ –њ—А–∞–≤—Л–є –ґ–µ–ї—Г–і–Њ—З–µ–Ї –≤¬†–≤–Є–і–µ —В—А–∞–±–µ–Ї—Г–ї—П—А–љ–Њ–є –Ј–Њ–љ—Л –Є¬†–≤—Л–љ–Њ—Б–љ–Њ–≥–Њ –Ї–∞–љ–∞–ї–∞. –Р—В—А–Є–∞–ї–Є–Ј–Њ–≤–∞–љ–љ–∞—П —З–∞—Б—В—М –ґ–µ–ї—Г–і–Њ—З–Ї–∞ (–≤—Е–Њ–і–љ–Њ–є –Ї–∞–љ–∞–ї) —А–∞—Б—И–Є—А–µ–љ–∞, –Љ–Њ–ґ–µ—В —Б–Њ—Б—В–∞–≤–ї—П—В—М –Њ—В¬†—В—А–µ—В–Є (—З–∞—Й–µ) –і–Њ –њ–Њ–ї–Њ–≤–Є–љ—Л (–≤¬†—В—П–ґ–µ–ї—Л—Е —Б–ї—Г—З–∞—П—Е) –Њ–±—К–µ–Љ–∞ –ґ–µ–ї—Г–і–Њ—З–Ї–∞. –†–∞—Б—И–Є—А—П–µ—В—Б—П –љ–µ¬†—В–Њ–ї—М–Ї–Њ –∞—В—А–Є–∞–ї–Є–Ј–Њ–≤–∞–љ–љ–∞—П —З–∞—Б—В—М, –љ–Њ¬†–Є —Д—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ–∞—П —З–∞—Б—В—М –њ—А–∞–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞. –Ю–љ–∞ –Љ–Њ–ґ–µ—В —Б–і–∞–≤–ї–Є–≤–∞—В—М –ї–µ–≤—Л–є –ґ–µ–ї—Г–і–Њ—З–µ–Ї –≤–њ–ї–Њ—В—М –і–Њ —Н–њ–Є–Ј–Њ–і–Њ–≤ –Њ–±—Б—В—А—Г–Ї—Ж–Є–Є –≤—Л–љ–Њ—Б–љ–Њ–≥–Њ —В—А–∞–Ї—В–∞ –ї–µ–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞ [11, 12] (—А–Є—Б.¬†1).
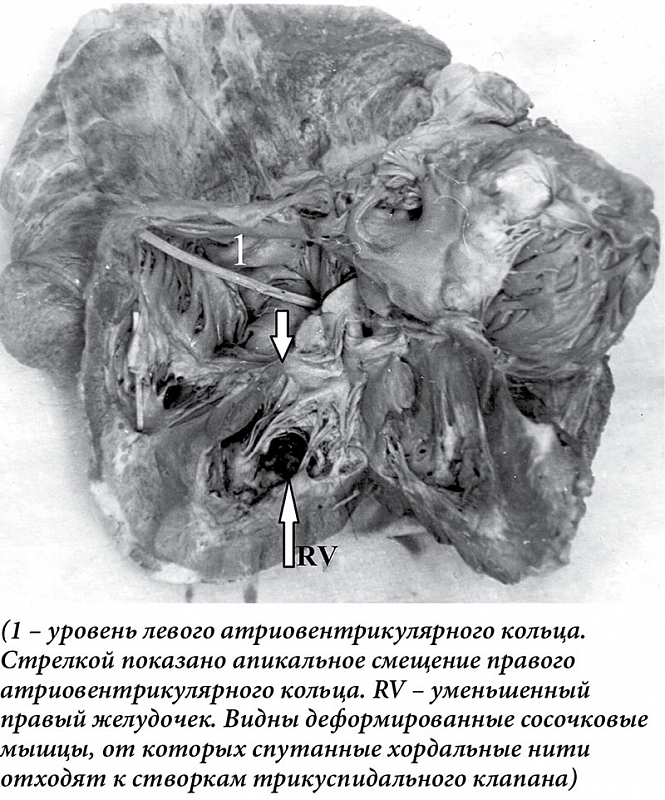
–Я–Њ¬†—А–µ–Ј—Г–ї—М—В–∞—В–∞–Љ –∞–љ–∞—В–Њ–Љ–Њ-–≥–Є—Б—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–≥–Њ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П, —Г¬†—В—А–µ—Е–Љ–µ—Б—П—З–љ–Њ–≥–Њ —А–µ–±–µ–љ–Ї–∞ –≤—Л—П–≤–ї–µ–љ–Њ —А–µ–Ј–Ї–Њ–µ —Г–≤–µ–ї–Є—З–µ–љ–Є–µ —А–∞–Ј–Љ–µ—А–∞ –Є¬†–Љ–∞—Б—Б—Л (50¬†–≥)¬†—Б–µ—А–і—Ж–∞. –Я—А–∞–≤–Њ–µ —Д–Є–±—А–Њ–Ј–љ–Њ–µ –∞—В—А–Є–Њ–≤–µ–љ—В—А–Є–Ї—Г–ї—П—А–љ–Њ–µ –Ї–Њ–ї—М—Ж–Њ —Б–Љ–µ—Й–µ–љ–Њ –≤–љ–Є–Ј –Є¬†–≤ —Б–∞–≥–Є—В—В–∞–ї—М–љ–Њ–є –њ–ї–Њ—Б–Ї–Њ—Б—В–Є, –µ–≥–Њ –њ—А–∞–≤–∞—П —Б—В–Њ—А–Њ–љ–∞ —А–∞—Б–њ–Њ–ї–Њ–ґ–µ–љ–∞ —Г¬†–≤–µ—А—Е—Г—И–Ї–Є, –∞¬†–Ј–∞–і–љ—П—П –Є¬†–њ–µ—А–µ–і–љ—П—П —Б—В–Њ—А–Њ–љ—Л¬†вАУ –≤–µ—А—В–Є–Ї–∞–ї—М–љ–Њ. –Я—А–∞–≤–Њ–µ –њ—А–µ–і—Б–µ—А–і–Є–µ –≤—Л–≥–ї—П–і–Є—В –Ї–∞–Ї –±–Њ–ї—М—И–∞—П —В–Њ–љ–Ї–Њ—Б—В–µ–љ–љ–∞—П –њ–Њ–ї–Њ—Б—В—М (5,5 √Ч 4,5 —Б–Љ) —Б¬†—В–Њ–ї—Й–Є–љ–Њ–є —Б—В–µ–љ–Ї–Є 0,1 —Б–Љ. –Э–∞¬†–Ј–∞–і–љ–µ–є —Б—В–µ–љ–Ї–µ –њ—А–∞–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞ –љ–∞—Е–Њ–і–Є—В—Б—П –љ–µ—А–Њ–≤–љ—Л–є —Н–љ–і–Њ–Ї–∞—А–і–Є–∞–ї—М–љ—Л–є –≤–∞–ї–Є–Ї, —А–∞—Б–њ–Њ–ї–Њ–ґ–µ–љ–љ—Л–є –≤–µ—А—В–Є–Ї–∞–ї—М–љ–Њ –Љ–µ–ґ–і—Г –Є—Б—В–Є–љ–љ–Њ–є –њ—А–µ–і—Б–µ—А–і–љ–Њ-–ґ–µ–ї—Г–і–Њ—З–Ї–Њ–≤–Њ–є¬†–≥—А–∞–љ–Є—Ж–µ–є –Є¬†–≤–µ—А—Е—Г—И–Ї–Њ–є —Б–µ—А–і—Ж–∞. –Т¬†—В–∞–Ї–Њ–Љ –ґ–µ –љ–∞–њ—А–∞–≤–ї–µ–љ–Є–Є –Ї¬†–њ–µ—А–µ–і–љ–µ–є —Б—В–µ–љ–Ї–µ –њ—А–∞–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞ –њ—А–Є–Ї—А–µ–њ–ї—П–µ—В—Б—П –µ–і–Є–љ—Б—В–≤–µ–љ–љ—Л–є –њ–µ—А–µ–і–љ–Є–є –њ–∞—А—Г—Б. –≠–љ–і–Њ–Ї–∞—А–і–Є–∞–ї—М–љ—Л–є –≤–∞–ї–Є–Ї –Є¬†–њ–µ—А–µ–і–љ–Є–є –њ–∞—А—Г—Б –і–µ–ї—П—В –њ—А–∞–≤—Л–є –ґ–µ–ї—Г–і–Њ—З–µ–Ї –љ–∞¬†–і–≤–µ —З–∞—Б—В–Є¬†вАУ –њ—А–∞–≤—Г—О, –љ–µ–њ–Њ—Б—А–µ–і—Б—В–≤–µ–љ–љ–Њ–µ –њ—А–Њ–і–Њ–ї–ґ–µ–љ–Є–µ –њ—А–∞–≤–Њ–≥–Њ –њ—А–µ–і—Б–µ—А–і–Є—П, —Б¬†—В–Њ–ї—Й–Є–љ–Њ–є —Б—В–µ–љ–Ї–Є 0,1 —Б–Љ –Є¬†—Г–Ј–Ї—Г—О –ї–µ–≤—Г—О¬†вАУ –≤—Л–љ–Њ—Б–љ–Њ–є —В—А–∞–Ї—В –ґ–µ–ї—Г–і–Њ—З–Ї–∞. –Т–љ—Г—В—А–µ–љ–љ—П—П –њ–Њ–≤–µ—А—Е–љ–Њ—Б—В—М –њ—А–∞–≤–Њ–≥–Њ –њ—А–µ–і—Б–µ—А–і–Є—П –Є¬†–∞—В—А–Є–∞–ї–Є–Ј–Њ–≤–∞–љ–љ–Њ–є —З–∞—Б—В–Є –њ—А–∞–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞¬†–≥–ї–∞–і–Ї–∞—П, –ї–Є—И–µ–љ–∞ —В—А–∞–±–µ–Ї—Г–ї—П—А–љ—Л—Е –Љ—Л—И—Ж. –Т¬†–Њ–±–ї–∞—Б—В–Є –≤–µ—А—Е—Г—И–Ї–Є –њ—А–∞–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞ —А–∞—Б–њ–Њ–ї–Њ–ґ–µ–љ—Л –і–≤–µ –Ї–Њ—А–Њ—В–Ї–Є–µ —В–Њ–љ–Ї–Є–µ —Б–Њ—Б–Њ—З–Ї–Њ–≤—Л–µ –Љ—Л—И—Ж—Л. –Ю—В¬†–Є—Е –≤–µ—А—Е—Г—И–µ–Ї –Њ—В—Е–Њ–і—П—В —В–Њ–љ–Ї–Є–µ —Е–Њ—А–і–∞–ї—М–љ—Л–µ –љ–Є—В–Є, –Ї–Њ—В–Њ—А—Л–µ –Ї—А–µ–њ—П—В—Б—П –Ї¬†–ї–µ–≤–Њ–є —Б—В–Њ—А–Њ–љ–µ –њ–µ—А–µ–і–љ–µ–≥–Њ –њ–∞—А—Г—Б–∞ –Є¬†–Њ–±—А–∞–Ј–Њ–≤—Л–≤–∞—О—В –љ–∞¬†–љ–µ–Љ –±–µ—Б–њ–Њ—А—П–і–Њ—З–љ—Л–µ —Б–њ–ї–µ—В–µ–љ–Є—П. –Ю–≤–∞–ї—М–љ–Њ–µ –Њ–Ї–љ–Њ –Њ—В–Ї—А—Л—В–Њ. –Ь–Є–Ї—А–Њ—Б–Ї–Њ–њ–Є—З–µ—Б–Ї–Є¬†вАУ –≤¬†–Љ–Є–Њ–Ї–∞—А–і–µ –њ—А–∞–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞ –≤—Л—А–∞–ґ–µ–љ–љ—Л–є –Њ—В–µ–Ї –Љ–µ–ґ–Љ—Л—И–µ—З–љ—Л—Е –њ—А–Њ—Б—В—А–∞–љ—Б—В–≤, –µ–і–Є–љ–Є—З–љ—Л–µ –Є–љ—Д–Є–ї—М—В—А–∞—В—Л –Є–Ј¬†–≥–Є—Б—В–Є–Њ—Ж–Є—В–Њ–≤. –°—Г–±—Н–љ–і–Њ–Ї–∞—А–і–Є–∞–ї—М–љ–Њ –Њ–±–љ–∞—А—Г–ґ–Є–≤–∞—О—В—Б—П –Љ–Є–Ї—А–Њ–љ–µ–Ї—А–Њ–Ј—Л.
–£¬†–Љ—Г–ґ—З–Є–љ—Л 62 –ї–µ—В, –Ј–∞–љ—П—В–Њ–≥–Њ —Д–Є–Ј–Є—З–µ—Б–Ї–Є–Љ —В—А—Г–і–Њ–Љ (–Ї–∞–Љ–µ–љ—Й–Є–Ї), –і–Њ—Б—В–∞–≤–ї–µ–љ–љ–Њ–≥–Њ –≤¬†—Б—В–∞—Ж–Є–Њ–љ–∞—А —Б¬†–Љ–∞–љ–Є—Д–µ—Б—В–љ—Л–Љ–Є –њ—А–Є–Ј–љ–∞–Ї–∞–Љ–Є —Б–µ—А–і–µ—З–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є, –њ—А–Є –∞—Г—В–Њ–њ—Б–Є–Є –Њ–±–љ–∞—А—Г–ґ–µ–љ–∞ –ґ–Є–і–Ї–Њ—Б—В—М –≤¬†—Б–µ—А–Њ–Ј–љ—Л—Е –њ–Њ–ї–Њ—Б—В—П—Е. –Ь–∞—Б—Б–∞ (470¬†–≥)¬†–Є —А–∞–Ј–Љ–µ—А—Л —Б–µ—А–і—Ж–∞ —Г–≤–µ–ї–Є—З–µ–љ—Л –Ј–∞ —Б—З–µ—В –њ—А–∞–≤—Л—Е –Њ—В–і–µ–ї–Њ–≤. –Я—А–∞–≤–Њ–µ —Д–Є–±—А–Њ–Ј–љ–Њ–µ –Ї–Њ–ї—М—Ж–Њ —Б–Љ–µ—Й–µ–љ–Њ –Ї¬†–≤–µ—А—Е—Г—И–Ї–µ –ґ–µ–ї—Г–і–Њ—З–Ї–∞. –Ъ¬†–Ї–Њ–ї—М—Ж—Г –њ—А–Є–Ї—А–µ–њ–ї—П—О—В—Б—П —В—А–Є –њ–∞—А—Г—Б–∞ —В—А–Є–Ї—Г—Б–њ–Є–і–∞–ї—М–љ–Њ–≥–Њ –Ї–ї–∞–њ–∞–љ–∞. –Я—А–∞–≤–Њ–µ –њ—А–µ–і—Б–µ—А–і–Є–µ —Г–≤–µ–ї–Є—З–µ–љ–Њ, —В–Њ–љ–Ї–Њ—Б—В–µ–љ–љ–Њ–µ. –Я—А–∞–≤—Л–є –ґ–µ–ї—Г–і–Њ—З–µ–Ї —Г–Љ–µ–љ—М—И–µ–љ, –Њ—В¬†—Д–Є–±—А–Њ–Ј–љ–Њ–≥–Њ –Ї–Њ–ї—М—Ж–∞ –і–Њ –≤–µ—А—Е—Г—И–Ї–Є¬†вАУ 3,2¬†—Б–Љ, —И–Є—А–Є–љ–∞ –ґ–µ–ї—Г–і–Њ—З–Ї–∞ –њ–Њ–і —Д–Є–±—А–Њ–Ј–љ—Л–Љ –Ї–Њ–ї—М—Ж–Њ–Љ¬†вАУ 3,6 —Б–Љ. –°—В–µ–љ–Ї–Є –њ—А–∞–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞ —Г—В–Њ–ї—Й–µ–љ—Л –і–Њ 1,5¬†–љ–∞¬†—Г—А–Њ–≤–љ–µ —Д–Є–±—А–Њ–Ј–љ–Њ–≥–Њ –Ї–Њ–ї—М—Ж–∞ –Є¬†3,5 —Б–Љ¬†вАУ –≤¬†–Њ–±–ї–∞—Б—В–Є –≤–µ—А—Е—Г—И–Ї–Є. –Т¬†–≤–µ—А—Е—Г—И–Ї–µ –ґ–µ–ї—Г–і–Њ—З–Ї–∞ –Є–Љ–µ—О—В—Б—П –і–≤–µ –Ї–Њ—А–Њ—В–Ї–Є–µ —В–Њ–ї—Б—В—Л–µ —Б–Њ—Б–Њ—З–Ї–Њ–≤—Л–µ –Љ—Л—И—Ж—Л (–њ–µ—А–µ–і–љ—П—П –Є¬†–Ј–∞–і–љ—П—П). –Ю—В¬†–њ–µ—А–µ–і–љ–µ–є –Љ—Л—И—Ж—Л —Е–Њ—А–і–∞–ї—М–љ—Л–µ –љ–Є—В–Є —В—П–љ—Г—В—Б—П –Ї¬†–њ–µ—А–µ–і–љ–µ–Љ—Г –њ–∞—А—Г—Б—Г —В—А–Є–Ї—Г—Б–њ–Є–і–∞–ї—М–љ–Њ–≥–Њ –Ї–ї–∞–њ–∞–љ–∞, –Њ—В¬†–Ј–∞–і–љ–µ–є¬†вАУ –Ї¬†–Ј–∞–і–љ–µ–Љ—Г –Є¬†–Љ–µ–і–Є–∞–ї—М–љ–Њ–Љ—Г. –•–Њ—А–і–∞–ї—М–љ—Л–µ –љ–Є—В–Є —В–Њ–љ–Ї–Є–µ, –Ї–Њ—А–Њ—В–Ї–Є–µ. –≠–љ–і–Њ–Ї–∞—А–і —Г—В–Њ–ї—Й–µ–љ, –±–µ–ї–µ—Б–Њ–≤–∞—В—Л–є. –§–Є–±—А–Њ–Ј–љ—Л–µ —В—П–ґ–Є –Њ—В¬†—Н–љ–і–Њ–Ї–∞—А–і–∞ –њ—А–Њ–љ–Є–Ї–∞—О—В –љ–∞¬†–≤—Б—О —В–Њ–ї—Й–Є–љ—Г –Љ–Є–Њ–Ї–∞—А–і–∞ –њ—А–∞–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞. –Т—Л—А–∞–ґ–µ–љ–∞¬†–≥–Є–њ–µ—А—В—А–Њ—Д–Є—П –Љ–µ–ґ–ґ–µ–ї—Г–і–Њ—З–Ї–Њ–≤–Њ–є –њ–µ—А–µ–≥–Њ—А–Њ–і–Ї–Є –Є¬†—Б—В–µ–љ–Ї–Є –ї–µ–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞. –У–Є—Б—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є¬†вАУ —Н–љ–і–Њ–Ї–∞—А–і –њ—А–∞–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞ —А–µ–Ј–Ї–Њ —Г—В–Њ–ї—Й–µ–љ –Ј–∞ —Б—З–µ—В —А–∞–Ј—А–∞—Б—В–∞–љ–Є—П –Ї–Њ–ї–ї–∞–≥–µ–љ–Њ–≤—Л—Е –≤–Њ–ї–Њ–Ї–Њ–љ. –Ь—Л—И–µ—З–љ—Л–µ –≤–Њ–ї–Њ–Ї–љ–∞ –њ—А–∞–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞¬†–≥–Є–њ–µ—А—В—А–Њ—Д–Є—А–Њ–≤–∞–љ—Л. –Ь–µ–ґ–і—Г –љ–Є–Љ–Є —И–Є—А–Њ–Ї–Є–µ –њ—А–Њ—Б–ї–Њ–є–Ї–Є –Ј—А–µ–ї–Њ–є —Б–Њ–µ–і–Є–љ–Є—В–µ–ї—М–љ–Њ–є —В–Ї–∞–љ–Є. –Ъ–Њ–ї–ї–∞–≥–µ–љ–Њ–≤—Л–µ –≤–Њ–ї–Њ–Ї–љ–∞ —А–∞—Б–њ–Њ–ї–Њ–ґ–µ–љ—Л –њ–∞—А–∞–ї–ї–µ–ї—М–љ–Њ, —Б—А–µ–і–Є –љ–Є—Е –љ–µ–Љ–љ–Њ–≥–Њ—З–Є—Б–ї–µ–љ–љ—Л–µ –Ї–ї–µ—В–Ї–Є —В–Є–њ–∞ —Д–Є–±—А–Њ–±–ї–∞—Б—В–Њ–≤. –®–Є—А–Њ–Ї–Є–µ —В—П–ґ–Є –Њ—В¬†—Н–љ–і–Њ–Ї–∞—А–і–∞¬†–≥–ї—Г–±–Њ–Ї–Њ –њ—А–Њ–љ–Є–Ї–∞—О—В –≤¬†–Љ–Є–Њ–Ї–∞—А–і, –Њ–±—А–∞–Ј—Г—П –њ–Њ–ї—П —Б–Ї–ї–µ—А–Њ–Ј–∞. –Т¬†–Њ—З–∞–≥–∞—Е —Б–Ї–ї–µ—А–Њ–Ј–∞¬†вАУ –∞—В—А–Њ—Д–Є—А–Њ–≤–∞–љ–љ—Л–µ –Љ—Л—И–µ—З–љ—Л–µ –њ—Г—З–Ї–Є (—А–Є—Б.¬†2).
–У–µ–Љ–Њ–і–Є–љ–∞–Љ–Є–Ї–∞ –Њ–њ—А–µ–і–µ–ї—П–µ—В—Б—П –≤—Л—А–∞–ґ–µ–љ–љ–Њ—Б—В—М—О –∞–љ–Њ–Љ–∞–ї–Є–Є. –Ь–Є–љ–Є–Љ–∞–ї—М–љ–Њ–µ —Б–Љ–µ—Й–µ–љ–Є–µ –њ—А–∞–≤–Њ–≥–Њ –∞—В—А–Є–Њ–≤–µ–љ—В—А–Є–Ї—Г–ї—П—А–љ–Њ–≥–Њ –Ї–Њ–ї—М—Ж–∞ –њ—А–Є —Г–Љ–µ—А–µ–љ–љ–Њ–є —А–µ–≥—Г—А–≥–Є—В–∞—Ж–Є–Є –Љ–Њ–ґ–µ—В –і–ї–Є—В–µ–ї—М–љ–Њ–µ –≤—А–µ–Љ—П –Њ—Б—В–∞–≤–∞—В—М—Б—П –±–µ–Ј –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є –Ј–љ–∞—З–Є–Љ—Л—Е –њ–Њ—Б–ї–µ–і—Б—В–≤–Є–є. –°—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ–µ —Б–Љ–µ—Й–µ–љ–Є–µ –Ї–ї–∞–њ–∞–љ–љ–Њ–≥–Њ –Ї–Њ–ї—М—Ж–∞ —Б¬†–≤—Л—А–∞–ґ–µ–љ–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М—О —В—А–Є–Ї—Г—Б–њ–Є–і–∞–ї—М–љ–Њ–≥–Њ –Ї–ї–∞–њ–∞–љ–∞, –Ј–љ–∞—З–Є—В–µ–ї—М–љ—Л–Љ —А–∞—Б—И–Є—А–µ–љ–Є–µ–Љ –њ–Њ–ї–Њ—Б—В–Є –њ—А–∞–≤–Њ–≥–Њ –њ—А–µ–і—Б–µ—А–і–Є—П –Є¬†—Б–Њ–Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ —Г–Љ–µ–љ—М—И–µ–љ–Є–µ–Љ –њ–Њ–ї–Њ—Б—В–Є –њ—А–∞–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞ –њ—А–Є–≤–Њ–і–Є—В –Ї¬†–і–µ–Ї–Њ–Љ–њ–µ–љ—Б–∞—Ж–Є–Є, –Є–љ–Њ–≥–і–∞ —Г–ґ–µ —Г¬†–њ–ї–Њ–і–∞. –†–∞–Ј–Љ–µ—А—Л —Д—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ–Њ –∞–Ї—В–Є–≤–љ–Њ–≥–Њ –њ—А–∞–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞ (–Ј–∞ –Є—Б–Ї–ї—О—З–µ–љ–Є–µ–Љ –µ–≥–Њ –∞—В—А–Є–∞–ї–Є–Ј–Њ–≤–∞–љ–љ–Њ–є —З–∞—Б—В–Є) –Љ–Њ–≥—Г—В –±—Л—В—М –љ–∞—Б—В–Њ–ї—М–Ї–Њ –Љ–∞–ї—Л, —З—В–Њ –љ–µ¬†—Б–њ–Њ—Б–Њ–±–љ—Л –Њ–±–µ—Б–њ–µ—З–Є—В—М –і–Њ—Б—В–∞—В–Њ—З–љ–Њ–µ —Б–Є—Б—В–Њ–ї–Є—З–µ—Б–Ї–Њ–µ –і–∞–≤–ї–µ–љ–Є–µ. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –Є–Љ–µ–µ—В –Љ–µ—Б—В–Њ –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М —В—А–µ—Е—Б—В–≤–Њ—А—З–∞—В–Њ–≥–Њ –Ї–ї–∞–њ–∞–љ–∞. –¶–Є–∞–љ–Њ–Ј –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ —В—А–µ–Љ—П –њ—А–Є—З–Є–љ–∞–Љ–Є:
- –і–µ—Д–µ–Ї—В–Њ–Љ –Љ–µ–ґ–њ—А–µ–і—Б–µ—А–і–љ–Њ–є –њ–µ—А–µ–≥–Њ—А–Њ–і–Ї–Є –Є–ї–Є –Њ—В–Ї—А—Л—В—Л–Љ –Њ–≤–∞–ї—М–љ—Л–Љ –Њ–Ї–љ–Њ–Љ;
- —Б–љ–Є–ґ–µ–љ–Є–µ–Љ –Ї—А–Њ–≤–Њ—В–Њ–Ї–∞ —З–µ—А–µ–Ј –ї–µ–≥–Ї–Є–µ –њ—А–Є —Д—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ–Њ–Љ –Є–ї–Є –∞–љ–∞—В–Њ–Љ–Є—З–µ—Б–Ї–Њ–Љ —Б—В–µ–љ–Њ–Ј–µ –Ї–ї–∞–њ–∞–љ–∞ –ї–µ–≥–Њ—З–љ–Њ–є –∞—А—В–µ—А–Є–Є;
- –≤—Л—Б–Њ–Ї–Є–Љ —Б–Њ–њ—А–Њ—В–Є–≤–ї–µ–љ–Є–µ–Љ —Б–Њ—Б—Г–і–Њ–≤ –ї–µ–≥–Ї–Њ–≥–Њ —Г¬†–љ–Њ–≤–Њ—А–Њ–ґ–і–µ–љ–љ—Л—Е.
–°–Њ—Б—В–Њ—П–љ–Є–µ –љ–µ—Б–Ї–Њ–ї—М–Ї–Њ —Б–Љ—П–≥—З–∞–µ—В –Њ—В–Ї—А—Л—В—Л–є –∞—А—В–µ—А–Є–∞–ї—М–љ—Л–є –њ—А–Њ—В–Њ–Ї, –њ–Њ¬†–Ї—А–∞–є–љ–µ–є –Љ–µ—А–µ –і–Њ —Б–љ–Є–ґ–µ–љ–Є—П —Б–Њ–њ—А–Њ—В–Є–≤–ї–µ–љ–Є—П –ї–µ–≥–Њ—З–љ—Л—Е —Б–Њ—Б—Г–і–Њ–≤ –њ—А–Є –њ–µ—А–µ—Б—В—А–Њ–є–Ї–µ –њ–ї–Њ–і–љ–Њ–≥–Њ —В–Є–њ–∞ –Ї—А–Њ–≤–Њ–Њ–±—А–∞—Й–µ–љ–Є—П. –Т¬†–і–∞–ї—М–љ–µ–є—И–µ–Љ –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ —А–∞–Ј–≤–Є—В–Є–µ —Б–µ—А–і–µ—З–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є –љ–µ¬†—В–Њ–ї—М–Ї–Њ –Є–Ј-–Ј–∞ –Љ–∞–ї–Њ–≥–Њ —А–∞–Ј–Љ–µ—А–∞ –њ—А–∞–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞ —Б¬†–Ї—А–∞–є–љ–µ –љ–Є–Ј–Ї–Є–Љ –Ї–Њ–Љ–њ–ї–∞–µ–љ—Б–Њ–Љ –Є¬†—А–µ–≥—Г—А–≥–Є—В–∞—Ж–Є–µ–є —З–µ—А–µ–Ј –њ—А–∞–≤–Њ–µ –∞—В—А–Є–Њ–≤–µ–љ—В—А–Є–Ї—Г–ї—П—А–љ–Њ–µ –Њ—В–≤–µ—А—Б—В–Є–µ, –љ–Њ¬†—В–∞–Ї–ґ–µ –Є–Ј-–Ј–∞ –і–Є—Б—Д—Г–љ–Ї—Ж–Є–Є –Є¬†–і–Є—Б—Е—А–Њ–љ–Є–Є –ї–µ–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞, —З–∞—Б—В–Њ –≤–Њ–Ј–љ–Є–Ї–∞—О—Й–µ–є —Б—Г–њ—А–∞–≤–µ–љ—В—А–Є–Ї—Г–ї—П—А–љ–Њ–є —В–∞—Е–Є–Ї–∞—А–і–Є–Є¬†вАУ 25вАУ50% –≤—Б–µ—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤. –£¬†–Љ–љ–Њ–≥–Є—Е –Є–Ј¬†–љ–Є—Е –Њ–±–љ–∞—А—Г–ґ–Є–≤–∞–µ—В—Б—П —Б–Є–љ–і—А–Њ–Љ –Т–Њ–ї—М—Д—Д–∞¬†вАУ –Я–∞—А–Ї–Є–љ—Б–Њ–љ–∞¬†вАУ –£–∞–є—В–∞ [13, 14].
–Ъ–ї–Є–љ–Є—З–µ—Б–Ї–∞—П –Ї–∞—А—В–Є–љ–∞
–Ъ–ї–Є–љ–Є—З–µ—Б–Ї–∞—П –Ї–∞—А—В–Є–љ–∞ –њ–Њ–ї–љ–Њ—Б—В—М—О –Њ–њ—А–µ–і–µ–ї—П–µ—В—Б—П —Б—В–µ–њ–µ–љ—М—О —Б–Љ–µ—Й–µ–љ–Є—П –њ—А–∞–≤–Њ–≥–Њ –∞—В—А–Є–Њ–≤–µ–љ—В—А–Є–Ї—Г–ї—П—А–љ–Њ–≥–Њ –Ї–Њ–ї—М—Ж–∞ –Є¬†–љ–∞–ї–Є—З–Є–µ–Љ —Б–Њ–њ—Г—В—Б—В–≤—Г—О—Й–Є—Е –њ–Њ—А–Њ–Ї–Њ–≤. –І–µ–Љ –Љ–µ–љ—М—И–µ —Б–Љ–µ—Й–µ–љ–Є–µ, —В–µ–Љ –њ–Њ–Ј–ґ–µ –Љ–∞–љ–Є—Д–µ—Б—В–Є—А—Г–µ—В –Ї–∞—А–і–Є–∞–ї—М–љ–∞—П —Б–Є–Љ–њ—В–Њ–Љ–∞—В–Є–Ї–∞. –Э–Њ—Б–Є—В–µ–ї–Є –њ–Њ—А–Њ–Ї–∞ –Љ–Њ–≥—Г—В –і–Њ–ґ–Є—В—М –і–Њ –Ј—А–µ–ї–Њ–≥–Њ –≤–Њ–Ј—А–∞—Б—В–∞ –±–µ–Ј —Е–Є—А—Г—А–≥–Є—З–µ—Б–Ї–Њ–≥–Њ –≤–Љ–µ—И–∞—В–µ–ї—М—Б—В–≤–∞ [15], —З—В–Њ –Ј–∞—Д–Є–Ї—Б–Є—А–Њ–≤–∞–љ–Њ –Є¬†–≤ –љ–∞—И–µ–Љ –љ–∞–±–ї—О–і–µ–љ–Є–Є. –£¬†–≤—Б–µ—Е –њ–∞—Ж–Є–µ–љ—В–Њ–≤, –љ–∞—Е–Њ–і–Є–≤—И–Є—Е—Б—П –њ–Њ–і –љ–∞—И–Є–Љ –љ–∞–±–ї—О–і–µ–љ–Є–µ–Љ, –Њ—В–Љ–µ—З–∞–ї–Є—Б—М —В–∞—Е–Є–Ї–∞—А–і–Є—П, —А–∞—Б—И–Є—А–µ–љ–Є–µ¬†–≥—А–∞–љ–Є—Ж —Б–µ—А–і—Ж–∞, –Њ–і—Л—И–Ї–∞, –Ј–∞—Б—В–Њ–є–љ—Л–µ —П–≤–ї–µ–љ–Є—П –≤¬†–ї–µ–≥–Ї–Є—Е –Є¬†—Г–≤–µ–ї–Є—З–µ–љ–Є–µ —А–∞–Ј–Љ–µ—А–Њ–≤ –њ–µ—З–µ–љ–Є. –Р—Г—Б–Ї—Г–ї—М—В–∞—В–Є–≤–љ–Њ –Њ–њ—А–µ–і–µ–ї—П–ї—Б—П¬†–≥—А—Г–±—Л–є —Б–Є—Б—В–Њ–ї–Є—З–µ—Б–Ї–Є–є —И—Г–Љ –≤¬†–Њ—Б–љ–Њ–≤–∞–љ–Є–Є —Б–µ—А–і—Ж–∞ –Є¬†—Б–Њ—Б—Г–і–Њ–≤. –†–µ–љ—В–≥–µ–љ–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є —Д–Є–Ї—Б–Є—А–Њ–≤–∞–ї–Є –Ї–∞—А–і–Є–Њ–Љ–µ–≥–∞–ї–Є—О, –ї–µ–≥–Њ—З–љ—Г—О –Њ–ї–Є–≥–µ–Љ–Є—О, —А–∞—Б—И–Є—А–µ–љ–Є–µ –њ—А–∞–≤–Њ–≥–Њ –њ—А–µ–і—Б–µ—А–і–Є—П –Є¬†–њ—А–∞–≤–Њ–≥–Њ —Б–Є–ї—Г—Н—В–∞ —Б–µ—А–і—Ж–∞.
–Ъ–ї–Є–љ–Є—З–µ—Б–Ї–∞—П –Ї–∞—А—В–Є–љ–∞ –∞–љ–Њ–Љ–∞–ї–Є–Є –≠–±—И—В–µ–є–љ–∞ –Ј–∞–≤–Є—Б–Є—В –љ–µ¬†—В–Њ–ї—М–Ї–Њ –Њ—В¬†—Б—В–µ–њ–µ–љ–Є —Б–Љ–µ—Й–µ–љ–Є—П –Ї–ї–∞–њ–∞–љ–∞, –љ–Њ¬†–Є –Њ—В¬†–≤–Њ–Ј—А–∞—Б—В–∞ –Љ–∞–љ–Є—Д–µ—Б—В–∞—Ж–Є–Є [16, 17]:
- –њ–ї–Њ–і: —В–Є–њ–Є—З–љ—Г—О —Н—Е–Њ–≥—А–∞—Д–Є—З–µ—Б–Ї—Г—О –Ї–∞—А—В–Є–љ—Г —Г–і–∞–µ—В—Б—П –Ј–∞—А–µ–≥–Є—Б—В—А–Є—А–Њ–≤–∞—В—М —Г¬†86% –њ–Њ—А–∞–ґ–µ–љ–љ—Л—Е, –∞—А–Є—В–Љ–Є—О¬†вАУ —Г¬†5%;
- –љ–Њ–≤–Њ—А–Њ–ґ–і–µ–љ–љ—Л–є (0вАУ1 –Љ–µ—Б—П—Ж): —Ж–Є–∞–љ–Њ–Ј —А–µ–≥–Є—Б—В—А–Є—А—Г–µ—В—Б—П —Г¬†74%, —Б–µ—А–і–µ—З–љ–∞—П –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М¬†вАУ —Г¬†10% (–Њ—В–Ї–∞–Ј –Њ—В¬†–µ–і—Л, –Ј–∞–і–µ—А–ґ–Ї–∞ —А–∞–Ј–≤–Є—В–Є—П), —И—Г–Љ –≤¬†—Б–µ—А–і—Ж–µ¬†вАУ —Г¬†9%;
- 2 –Љ–µ—Б—П—Ж–∞¬†вАУ 2¬†–≥–Њ–і–∞: —Ж–Є–∞–љ–Њ–Ј¬†вАУ 35% —Б–ї—Г—З–∞–µ–≤, —Б–µ—А–і–µ—З–љ–∞—П –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М¬†вАУ 43%,¬† —И—Г–Љ –≤¬†—Б–µ—А–і—Ж–µ¬†вАУ 13%;
- 3¬†–≥–Њ–і–∞¬†вАУ 10 –ї–µ—В: —Ж–Є–∞–љ–Њ–Ј¬†вАУ 14%, —Б–µ—А–і–µ—З–љ–∞—П –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М¬†вАУ 8%, –∞—А–Є—В–Љ–Є—П¬†вАУ 12%, —И—Г–Љ –≤¬†—Б–µ—А–і—Ж–µ¬†вАУ 66%;
- 11вАУ18 –ї–µ—В: —Ж–Є–∞–љ–Њ–Ј¬†вАУ 13%, —Б–µ—А–і–µ—З–љ–∞—П –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М¬†вАУ 13%, –∞—А–Є—В–Љ–Є—П¬†вАУ 40%, —И—Г–Љ –≤¬†—Б–µ—А–і—Ж–µ¬†вАУ 33%;
- –≤–Ј—А–Њ—Б–ї—Л–µ (—Б—В–∞—А—И–µ 18 –ї–µ—В): —Ж–Є–∞–љ–Њ–Ј¬†вАУ 4%, —Б–µ—А–і–µ—З–љ–∞—П –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М¬†вАУ 26%, –∞—А–Є—В–Љ–Є—П¬†вАУ 43%, —И—Г–Љ –≤¬†—Б–µ—А–і—Ж–µ¬†вАУ 13%, –±–Њ–ї—М –≤¬†–Њ–±–ї–∞—Б—В–Є —Б–µ—А–і—Ж–∞¬†вАУ 20%, –Њ–±–Љ–Њ—А–Њ–Ї¬†вАУ 6%.
–Я—А–Є –∞–љ–Њ–Љ–∞–ї–Є–Є –≠–±—И—В–µ–є–љ–∞ —Н–ї–µ–Ї—В—А–Њ–Ї–∞—А–і–Є–Њ–≥—А–∞–Љ–Љ–∞ (–≠–Ъ–У) –њ–Њ–Ї–∞–Ј—Л–≤–∞–µ—В –Њ—Б—В—А—Г—О –≤—Л—Б–Њ–Ї—Г—О –≤–Њ–ї–љ—Г –† (–њ—А–Є–Ј–љ–∞–Ї —А–∞—Б—И–Є—А–µ–љ–Є—П –њ—А–∞–≤–Њ–≥–Њ –њ—А–µ–і—Б–µ—А–і–Є—П), –Њ—В–Ї–ї–Њ–љ–µ–љ–Є–µ —Н–ї–µ–Ї—В—А–Є—З–µ—Б–Ї–Њ–є –Њ—Б–Є —Б–µ—А–і—Ж–∞ –≤–њ—А–∞–≤–Њ, –±–ї–Њ–Ї–∞–і—Г –њ—А–∞–≤–Њ–є –љ–Њ–ґ–Ї–Є –њ—Г—З–Ї–∞ –У–Є—Б–∞. –Я—А–Є –љ–∞–ї–Є—З–Є–Є —Б–Є–љ–і—А–Њ–Љ–∞ –Т–Њ–ї—М—Д—Д–∞¬†вАУ –Я–∞—А–Ї–Є–љ—Б–Њ–љ–∞¬†вАУ –£–∞–є—В–∞¬†вАУ –Ї–Њ—А–Њ—В–Ї–Є–є –Є–љ—В–µ—А–≤–∞–ї PR¬†–Є¬†–≤–Њ–ї–љ—Г –і–µ–ї—М—В–∞. –Э–µ¬†–Є—Б–Ї–ї—О—З–µ–љ–∞ –њ—А–µ–і—Б–µ—А–і–љ–∞—П –∞—А–Є—В–Љ–Є—П.
–≠—Е–Њ–Ї–∞—А–і–Є–Њ–≥—А–∞—Д–Є—П¬†вАУ —А–µ—И–∞—О—Й–∞—П –Љ–µ—В–Њ–і–Є–Ї–∞ –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–Є. –¶–µ–ї—М —Г–ї—М—В—А–∞–Ј–≤—Г–Ї–Њ–≤–Њ–≥–Њ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П¬†вАУ –Њ–њ–Є—Б–∞—В—М –∞–љ–∞—В–Њ–Љ–Є—З–µ—Б–Ї–Є–µ —Е–∞—А–∞–Ї—В–µ—А–Є—Б—В–Є–Ї–Є –њ–Њ—А–Њ–Ї–∞, —Б–Њ–њ—Г—В—Б—В–≤—Г—О—Й–Є—Е –∞–љ–Њ–Љ–∞–ї–Є–є –Є¬†–≥–µ–Љ–Њ–і–Є–љ–∞–Љ–Є—З–µ—Б–Ї–Є–µ –љ–∞—А—Г—И–µ–љ–Є—П.
–Т¬†–Ь-—А–µ–ґ–Є–Љ–µ —В—А–Є–Ї—Г—Б–њ–Є–і–∞–ї—М–љ—Л–є –Ї–ї–∞–њ–∞–љ –ї–Њ—Ж–Є—А—Г–µ—В—Б—П –ї–µ–≤–µ–µ –Њ–±—Л—З–љ–Њ–≥–Њ. –Я–µ—А–µ–і–љ—П—П —Б—В–≤–Њ—А–Ї–∞ –Ї–∞–ґ–µ—В—Б—П –∞–љ–Њ–Љ–∞–ї—М–љ–Њ –±–Њ–ї—М—И–Њ–є. –Р–Љ–њ–ї–Є—В—Г–і–∞ –і–≤–Є–ґ–µ–љ–Є—П –Ї–ї–∞–њ–∞–љ–∞ —Г–≤–µ–ї–Є—З–µ–љ–∞. –Ч–∞–Ї—А—Л—В–Є–µ —Н—В–Њ–≥–Њ –Ї–ї–∞–њ–∞–љ–∞ –≤¬†–Њ—В–ї–Є—З–Є–µ –Њ—В¬†–Љ–Є—В—А–∞–ї—М–љ–Њ–≥–Њ –њ—А–Њ–Є—Б—Е–Њ–і–Є—В –њ–Њ–Ј–і–љ–µ–µ, —З—В–Њ –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–Њ –∞—В—А–Є–∞–ї–Є–Ј–∞—Ж–Є–µ–є –њ—А–∞–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞ –Є¬†—Б–љ–Є–ґ–µ–љ–Є–µ–Љ –µ–≥–Њ –љ–∞—Б–Њ—Б–љ–Њ–є —Д—Г–љ–Ї—Ж–Є–Є. –†–∞–Ј–љ–Є—Ж–∞ 40 –Љ—Б –Љ–µ–ґ–і—Г —В–Њ—З–Ї–∞–Љ–Є –Ј–∞–Ї—А—Л—В–Є—П —В—А–Є–Ї—Г—Б–њ–Є–і–∞–ї—М–љ–Њ–≥–Њ –Є¬†–Љ–Є—В—А–∞–ї—М–љ–Њ–≥–Њ –Ї–ї–∞–њ–∞–љ–Њ–≤¬†вАУ –і–Є–∞–≥–љ–Њ—Б—В–Є—З–µ—Б–Ї–Є –≤–∞–ґ–љ—Л–є –њ—А–Є–Ј–љ–∞–Ї –∞–љ–Њ–Љ–∞–ї–Є–Є –≠–±—И—В–µ–є–љ–∞, —Е–Њ—В—П –Є¬†–љ–µ –≤—Б–µ–≥–і–∞ —А–µ–≥–Є—Б—В—А–Є—А—Г–µ—В—Б—П. –Я—А–Є –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ—Б—В–Є –Њ–і–љ–Њ–Љ–Њ–Љ–µ–љ—В–љ–Њ–є —А–µ–≥–Є—Б—В—А–∞—Ж–Є–Є –Ї–ї–∞–њ–∞–љ–Њ–≤ –ї–µ–≥–Њ—З–љ–Њ–є –∞—А—В–µ—А–Є–Є –Є¬†–∞–Њ—А—В—Л –≤–Є–і–љ–Њ, —З—В–Њ –Ї–ї–∞–њ–∞–љ –ї–µ–≥–Њ—З–љ–Њ–є –∞—А—В–µ—А–Є–Є –Њ—В–Ї—А—Л–≤–∞–µ—В—Б—П –Є¬†–Ј–∞–Ї—А—Л–≤–∞–µ—В—Б—П –њ–Њ–Ј–ґ–µ, —З–µ–Љ –∞–Њ—А—В–∞–ї—М–љ—Л–є, —З—В–Њ —В–∞–Ї–ґ–µ –Њ—В—А–∞–ґ–∞–µ—В —Б–љ–Є–ґ–µ–љ–љ—Г—О –љ–∞—Б–Њ—Б–љ—Г—О —Д—Г–љ–Ї—Ж–Є—О –њ—А–∞–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞. –Э–∞–Є–±–Њ–ї–µ–µ –Є–љ—Д–Њ—А–Љ–∞—В–Є–≤–љ–∞ 2D-—Н—Е–Њ–Ї–∞—А–і–Є–Њ–≥—А–∞—Д–Є—П, —Б—З–Є—В–∞—О—Й–∞—П—Б—П –Ј–Њ–ї–Њ—В—Л–Љ —Б—В–∞–љ–і–∞—А—В–Њ–Љ –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–Є —Н—В–Њ–≥–Њ –њ–Њ—А–Њ–Ї–∞. –У–µ–Љ–Њ–і–Є–љ–∞–Љ–Є—З–µ—Б–Ї–Є–µ —Е–∞—А–∞–Ї—В–µ—А–Є—Б—В–Є–Ї–Є –Љ–Њ–ґ–љ–Њ –њ–Њ–ї—Г—З–Є—В—М –њ–Њ¬†—А–µ–Ј—Г–ї—М—В–∞—В–∞–Љ —Б–њ–µ–Ї—В—А–∞–ї—М–љ–Њ–є –Є¬†—Ж–≤–µ—В–љ–Њ–є –і–Њ–њ–њ–ї–µ—А–Њ–≥—А–∞—Д–Є–Є. –Ю–њ—В–Є–Љ–∞–ї—М–љ—Л —В—А–∞–љ—Б—В–Њ—А–∞–Ї–∞–ї—М–љ–Њ–µ —З–µ—В—Л—А–µ—Е–Ї–∞–Љ–µ—А–љ–Њ–µ —Б–Ї–∞–љ–Є—А–Њ–≤–∞–љ–Є–µ, —Б–Ї–∞–љ–Є—А–Њ–≤–∞–љ–Є–µ –њ–Њ¬†–њ–∞—А–∞—Б—В–µ—А–љ–∞–ї—М–љ—Л–Љ –Є¬†—Б—Г–±–Ї–Њ—Б—В–∞–ї—М–љ—Л–Љ –і–ї–Є–љ–љ–Њ–є –Є¬†–Ї–Њ—А–Њ—В–Ї–Њ–є –Њ—Б—П–Љ. –Я—А–Є –Њ–±—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є –њ–ї–Њ–і–∞ –њ–∞—В–Њ–ї–Њ–≥–Є—П —В—А–Є–Ї—Г—Б–њ–Є–і–∞–ї—М–љ–Њ–≥–Њ –Ї–ї–∞–њ–∞–љ–∞ –ї—Г—З—И–µ –≤—Б–µ–≥–Њ –≤—Л—П–≤–ї—П–µ—В—Б—П –њ—А–Є —З–µ—В—Л—А–µ—Е–Ї–∞–Љ–µ—А–љ–Њ–Љ —Б–Ї–∞–љ–Є—А–Њ–≤–∞–љ–Є–Є. –І—А–µ—Б–њ–Є—Й–µ–≤–Њ–і–љ–∞—П —Н—Е–Њ–≥—А–∞—Д–Є—П –Њ–њ—А–∞–≤–і–∞–љ–љ–∞ –њ—А–Є –Њ–њ–µ—А–∞—В–Є–≤–љ–Њ–Љ –≤–Љ–µ—И–∞—В–µ–ї—М—Б—В–≤–µ.
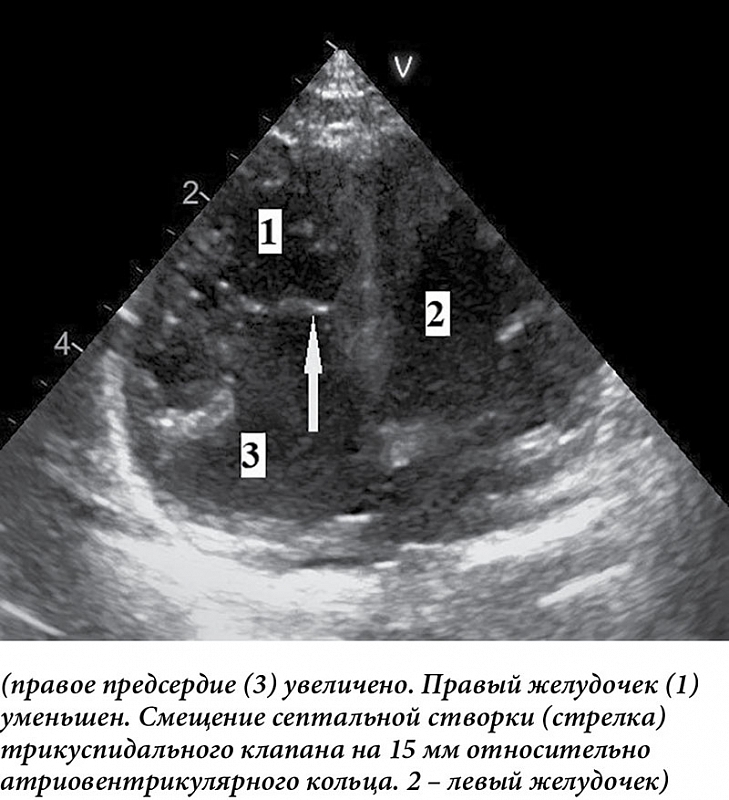
–Р–њ–Є–Ї–∞–ї—М–љ–Њ–µ —Б–Љ–µ—Й–µ–љ–Є–µ —Б–µ–њ—В–∞–ї—М–љ–Њ–є —Б—В–≤–Њ—А–Ї–Є (–њ—А–Є —В—П–ґ–µ–ї—Л—Е –≤–∞—А–Є–∞–љ—В–∞—Е –∞–љ–Њ–Љ–∞–ї–Є–Є) –Њ—В–љ–Њ—Б–Є—В–µ–ї—М–љ–Њ –Љ–Є—В—А–∞–ї—М–љ–Њ–≥–Њ –Ї–ї–∞–њ–∞–љ–∞ —Е–Њ—А–Њ—И–Њ –Ј–∞–Љ–µ—В–љ–Њ –љ–∞¬†—З–µ—В—Л—А–µ—Е–Ї–∞–Љ–µ—А–љ–Њ–Љ –Є–Ј–Њ–±—А–∞–ґ–µ–љ–Є–Є (—А–Є—Б.¬†3).
–£–≤–µ–ї–Є—З–µ–љ–Є–µ –њ—А–∞–≤–Њ–≥–Њ –њ—А–µ–і—Б–µ—А–і–Є—П –Њ–њ—В–Є–Љ–∞–ї—М–љ–Њ –Њ—Ж–µ–љ–Є–≤–∞—В—М –њ—А–Є —З–µ—В—Л—А–µ—Е–Ї–∞–Љ–µ—А–љ–Њ–Љ —Б–Ї–∞–љ–Є—А–Њ–≤–∞–љ–Є–Є, –њ–Њ¬†–њ–∞—А–∞—Б—В–µ—А–љ–∞–ї—М–љ–Њ–є –Ї–Њ—А–Њ—В–Ї–Њ–є –Њ—Б–Є –Є¬†—Б—Г–±–Ї–Њ—Б—В–∞–ї—М–љ–Њ. –Ґ—А–Є–Ї—Г—Б–њ–Є–і–∞–ї—М–љ—Л–є –Ї–ї–∞–њ–∞–љ –ї—Г—З—И–µ –≤—Б–µ–≥–Њ –ї–Њ—Ж–Є—А—Г–µ—В—Б—П –њ–Њ¬†–њ–∞—А–∞—Б—В–µ—А–љ–∞–ї—М–љ–Њ–є –і–ї–Є–љ–љ–Њ–є –Є¬†–Ї–Њ—А–Њ—В–Ї–Њ–є –Њ—Б—П–Љ, –њ—А–Є —З–µ—В—Л—А–µ—Е–Ї–∞–Љ–µ—А–љ–Њ–Љ –Є¬†—Б—Г–±–Ї–Њ—Б—В–∞–ї—М–љ–Њ–Љ —Б–Ї–∞–љ–Є—А–Њ–≤–∞–љ–Є–Є. –Я–Њ¬†–Є—В–Њ–≥–∞–Љ —В–∞–Ї–Њ–≥–Њ —Б–Ї–∞–љ–Є—А–Њ–≤–∞–љ–Є—П –Љ–Њ–ґ–љ–Њ –Њ–њ—А–µ–і–µ–ї–Є—В—М —Б—В–µ–њ–µ–љ—М —В—П–ґ–µ—Б—В–Є –∞–љ–Њ–Љ–∞–ї–Є–Є, —З—В–Њ –≤–∞–ґ–љ–Њ –і–ї—П –Њ–њ—А–µ–і–µ–ї–µ–љ–Є—П —Б—А–Њ–Ї–Њ–≤ –Њ–њ–µ—А–∞—В–Є–≤–љ–Њ–≥–Њ –≤–Љ–µ—И–∞—В–µ–ї—М—Б—В–≤–∞. –Ф–ї—П —Н—В–Њ–≥–Њ –њ—А–Є —З–µ—В—Л—А–µ—Е–Ї–∞–Љ–µ—А–љ–Њ–Љ —Б–Ї–∞–љ–Є—А–Њ–≤–∞–љ–Є–Є –≤¬†–Ї–Њ–љ—Ж–µ –і–Є–∞—Б—В–Њ–ї—Л –Њ–њ—А–µ–і–µ–ї—П—О—В —З–∞—Б—В–љ–Њ–µ –Њ—В¬†–і–µ–ї–µ–љ–Є—П —Б—Г–Љ–Љ—Л –њ–ї–Њ—Й–∞–і–Є –њ—А–∞–≤–Њ–≥–Њ –њ—А–µ–і—Б–µ—А–і–Є—П (RA) –Є¬†–∞—В—А–Є–∞–ї–Є–Ј–Њ–≤–∞–љ–љ–Њ–є —З–∞—Б—В–Є –њ—А–∞–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞ (aRV) –љ–∞¬†—Б—Г–Љ–Љ—Г –њ–ї–Њ—Й–∞–і–µ–є —Д—Г–љ–Ї—Ж–Є–Њ–љ–∞–ї—М–љ–Њ–є —З–∞—Б—В–Є –њ—А–∞–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞ (RV), –ї–µ–≤–Њ–≥–Њ –њ—А–µ–і—Б–µ—А–і–Є—П (LA) –Є¬†–ї–µ–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞ (LV): (RA + aRV)/(RV + LA + LV). –Х—Б–ї–Є –њ–Њ–ї—Г—З–µ–љ–љ–∞—П –≤–µ–ї–Є—З–Є–љ–∞ –Љ–µ–љ–µ–µ 0,5,¬†–≥–Њ–≤–Њ—А—П—В –Њ¬†–њ–µ—А–≤–Њ–є —Б—В–µ–њ–µ–љ–Є —В—П–ґ–µ—Б—В–Є (–ї–µ—В–∞–ї—М–љ–Њ—Б—В—М 0%). –Ч–љ–∞—З–µ–љ–Є–µ —Г–Ї–∞–Ј–∞–љ–љ–Њ–є –≤–µ–ї–Є—З–Є–љ—Л 0,5вАУ0,99 —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г–µ—В –Њ¬†–≤—В–Њ—А–Њ–є —Б—В–µ–њ–µ–љ–Є (–ї–µ—В–∞–ї—М–љ–Њ—Б—В—М –і–Њ 10%), 1вАУ1,49¬†вАУ –Њ¬†—В—А–µ—В—М–µ–є —Б—В–µ–њ–µ–љ–Є (–ї–µ—В–∞–ї—М–љ–Њ—Б—В—М 44%), –±–Њ–ї–µ–µ 1,5¬†вАУ —З–µ—В–≤–µ—А—В–Њ–є —Б—В–µ–њ–µ–љ–Є (–ї–µ—В–∞–ї—М–љ–Њ—Б—В—М –њ—А–∞–Ї—В–Є—З–µ—Б–Ї–Є 100%). –Ґ—А–µ—В—М—П –Є¬†—З–µ—В–≤–µ—А—В–∞—П —Б—В–µ–њ–µ–љ–Є —В—П–ґ–µ—Б—В–Є¬†вАУ –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–Є –≤—Л—Б–Њ–Ї–Њ–є –≤–µ—А–Њ—П—В–љ–Њ—Б—В–Є –ї–µ—В–∞–ї—М–љ–Њ–≥–Њ –Є—Б—Е–Њ–і–∞ [18вАУ20]. –Т¬†–і–∞–ї—М–љ–µ–є—И–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П—Е –±—Л–ї–Њ –њ–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ –љ–µ¬†–Љ–µ–љ—М—И–µ–µ –Ј–љ–∞—З–µ–љ–Є–µ –≤¬†–Ї–∞—З–µ—Б—В–≤–µ –њ—А–µ–і–Є–Ї—В–Њ—А–Њ–≤ –љ–µ–±–ї–∞–≥–Њ–њ—А–Є—П—В–љ—Л—Е –Є—Б—Е–Њ–і–Њ–≤ –Є–Љ–µ—О—В –і–Є—Б—В—А–µ—Б—Б –њ–ї–Њ–і–∞, –∞—В—А–µ–Ј–Є—П/—Б—В–µ–љ–Њ–Ј –ї–µ–≥–Њ—З–љ–Њ–є –∞—А—В–µ—А–Є–Є [21].
–Я–µ–і–Є–∞—В—А–Є—З–µ—Б–Ї–Є–µ –њ—А–Њ–±–ї–µ–Љ—Л –≤–µ–і–µ–љ–Є—П –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–∞–љ–Њ–Љ–∞–ї–Є–µ–є –≠–±—И—В–µ–є–љ–∞
–Ш–Ј¬†—Б–њ–µ—Ж–Є–∞–ї–Є—Б—В–Њ–≤ —Б¬†—А–µ–±–µ–љ–Ї–Њ–Љ —З–∞—Й–µ –≤—Б–µ–≥–Њ –Ї–Њ–љ—В–∞–Ї—В–Є—А—Г–µ—В –њ–µ–і–Є–∞—В—А. –Я–Њ—Н—В–Њ–Љ—Г –Є–Љ–µ–љ–љ–Њ –Њ—В¬†–љ–µ–≥–Њ –Ј–∞–≤–Є—Б–Є—В, –≤¬†–Ї–∞–Ї–Є–µ —Б—А–Њ–Ї–Є, –Ї¬†–Ї–∞–Ї–Њ–Љ—Г —Б–њ–µ—Ж–Є–∞–ї–Є—Б—В—Г, –љ–∞¬†–Ї–Њ–љ—Б—Г–ї—М—В–∞—Ж–Є—О –Є–ї–Є –і–Є—Б–њ–∞–љ—Б–µ—А–љ–Њ–µ –љ–∞–±–ї—О–і–µ–љ–Є–µ –±—Г–і–µ—В –љ–∞–њ—А–∞–≤–ї–µ–љ —А–µ–±–µ–љ–Њ–Ї. –Я–µ–і–Є–∞—В—А —А–µ—И–∞–µ—В –њ—А–Њ–±–ї–µ–Љ—Л –Љ–µ–ґ–і–Є—Б—Ж–Є–њ–ї–Є–љ–∞—А–љ–Њ–≥–Њ –Є¬†–Љ–µ–ґ–њ—А–Њ—Д–µ—Б—Б–Є–Њ–љ–∞–ї—М–љ–Њ–≥–Њ –≤–Ј–∞–Є–Љ–Њ–і–µ–є—Б—В–≤–Є—П.
–С–µ—Б—Б–Є–Љ–њ—В–Њ–Љ–љ—Л–µ –њ–∞—Ж–Є–µ–љ—В—Л —Б¬†–∞–љ–Њ–Љ–∞–ї–Є–µ–є –≠–±—И—В–µ–є–љ–∞ —Б¬†–Љ–Є–љ–Є–Љ–∞–ї—М–љ–Њ–є —А–µ–≥—Г—А–≥–Є—В–∞—Ж–Є–µ–є –≤¬†–Њ—В—Б—Г—В—Б—В–≤–Є–µ –і—А—Г–≥–Є—Е –њ–Њ—А–Њ–Ї–Њ–≤ —А–∞–Ј–≤–Є—В–Є—П –Є¬†–љ–∞—А—Г—И–µ–љ–Є—П —А–Є—В–Љ–∞ —Б–µ—А–і—Ж–∞ –Љ–Њ–≥—Г—В –љ–∞–±–ї—О–і–∞—В—М—Б—П –∞–Љ–±—Г–ї–∞—В–Њ—А–љ–Њ. –Т¬†–Њ—В—Б—Г—В—Б—В–≤–Є–µ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е –њ–Њ–Ї–∞–Ј–∞–љ–Є–є (—Б–љ–Є–ґ–µ–љ–Є–µ –∞–њ–њ–µ—В–Є—В–∞ –Є¬†—Д–Є–Ј–Є—З–µ—Б–Ї–Њ–є –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є, –њ–ї–Њ—Б–Ї–∞—П –≤–µ—Б–Њ–≤–∞—П –Ї—А–Є–≤–∞—П –Є–ї–Є –њ–µ—А–µ—Е–Њ–і –њ–Њ–Ї–∞–Ј–∞—В–µ–ї–µ–є –Љ–∞—Б—Б—Л —В–µ–ї–∞ –Є¬†—А–Њ—Б—В–∞ –љ–∞¬†–љ–Є–ґ–љ—О—О –њ–µ—А—Ж–µ–љ—В–Є–ї—М, –Њ–і—Л—И–Ї–∞) –Њ–і–Є–љ —А–∞–Ј –≤¬†3вАУ6 –Љ–µ—Б—П—Ж–µ–≤ –≤—Л–њ–Њ–ї–љ—П–µ—В—Б—П –≠–Ъ–У, –Њ–і–Є–љ —А–∞–Ј –≤¬†—И–µ—Б—В—М –Љ–µ—Б—П—Ж–µ–≤¬†вАУ —Н—Е–Њ–Ї–∞—А–і–Є–Њ–≥—А–∞—Д–Є—П –Є¬†–і–Њ–њ–њ–ї–µ—А–Њ–≥—А–∞—Д–Є—П, –Њ–њ—А–µ–і–µ–ї–µ–љ–Є–µ —Б–∞—В—Г—А–∞—Ж–Є–Є –Ї–Є—Б–ї–Њ—А–Њ–і–∞. –Ъ–Њ–љ—Б—Г–ї—М—В–∞—Ж–Є—П –Ї–∞—А–і–Є–Њ–ї–Њ–≥–∞ –њ—А–Њ–≤–Њ–і–Є—В—Б—П –Њ–і–Є–љ —А–∞–Ј –≤¬†—И–µ—Б—В—М –Љ–µ—Б—П—Ж–µ–≤, —А–µ–љ—В–≥–µ–љ–Њ–≥—А–∞—Д–Є—П¬†вАУ –њ–Њ¬†–њ–Њ–Ї–∞–Ј–∞–љ–Є—П–Љ.
–Т¬†—Б–ї—Г—З–∞–µ –њ—А–Є—Б–Њ–µ–і–Є–љ–µ–љ–Є—П –≤–Є—А—Г—Б–љ–Њ–є –Є/–Є–ї–Є –±–∞–Ї—В–µ—А–Є–∞–ї—М–љ–Њ–є –Є–љ—Д–µ–Ї—Ж–Є–Є –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ –Њ—Ж–µ–љ–Є—В—М –њ–Њ–Ї–∞–Ј–∞–љ–Є—П –Ї¬†–њ—А–Њ–≤–µ–і–µ–љ–Є—О –≠–Ъ–У –Є¬†—Г–ї—М—В—А–∞–Ј–≤—Г–Ї–Њ–≤–Њ–≥–Њ –Њ–±—Б–ї–µ–і–Њ–≤–∞–љ–Є—П —Б–µ—А–і—Ж–∞.
–Я—А–Њ—Д–Є–ї–∞–Ї—В–Є–Ї–∞ –±–∞–Ї—В–µ—А–Є–∞–ї—М–љ–Њ–≥–Њ —Н–љ–і–Њ–Ї–∞—А–і–Є—В–∞ –Њ—Б—Г—Й–µ—Б—В–≤–ї—П–µ—В—Б—П –њ–Њ–ґ–Є–Ј–љ–µ–љ–љ–Њ, –≤–∞–Ї—Ж–Є–љ–∞—Ж–Є—П¬†вАУ –њ–Њ¬†–Њ–±—Й–µ–Љ—Г¬†–≥—А–∞—Д–Є–Ї—Г. –Ю–±—П–Ј–∞—В–µ–ї—М–љ–∞ –≤–∞–Ї—Ж–Є–љ–∞—Ж–Є—П –Њ—В¬†–њ–љ–µ–≤–Љ–Њ- –Є¬†–Љ–µ–љ–Є–љ–≥–Њ–Ї–Њ–Ї–Ї–∞.
–Ф–Є–µ—В–∞¬†вАУ –±–µ–Ј –Њ—Б–Њ–±–µ–љ–љ–Њ—Б—В–µ–є. –Я—А–Є —В—П–ґ–µ–ї—Л—Е –≤–∞—А–Є–∞–љ—В–∞—Е –∞–љ–Њ–Љ–∞–ї–Є–Є –Є¬†–њ—А–Є–Ј–љ–∞–Ї–∞—Е —Б–µ—А–і–µ—З–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є –љ–∞–Ј–љ–∞—З–∞—О—В –≤—Л—Б–Њ–Ї–Њ–Ї–∞–ї–Њ—А–Є–є–љ—Л–µ —Б–Љ–µ—Б–Є. –§–Є–Ј–Є—З–µ—Б–Ї–∞—П –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –њ—А–Є –∞–љ–Њ–Љ–∞–ї–Є–Є –≠–±—И—В–µ–є–љ–∞ –Ј–∞–≤–Є—Б–Є—В –Њ—В¬†—Б—В–µ–њ–µ–љ–Є —Б–Љ–µ—Й–µ–љ–Є—П —Б—В–≤–Њ—А–Њ–Ї –Ї–ї–∞–њ–∞–љ–∞, —Б–Њ–њ—Г—В—Б—В–≤—Г—О—Й–Є—Е –њ–Њ—А–Њ–Ї–Њ–≤ –Є–ї–Є –љ–∞—А—Г—И–µ–љ–Є–є —А–Є—В–Љ–∞ —Б–µ—А–і—Ж–∞. –Х—Б–ї–Є —Б–Љ–µ—Й–µ–љ–Є–µ —Б—В–≤–Њ—А–Њ–Ї –љ–µ–Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ–µ, –љ–µ—В –њ–∞—А–Њ–Ї—Б–Є–Ј–Љ–∞–ї—М–љ–Њ–є —Б—Г–њ—А–∞–≤–µ–љ—В—А–Є–Ї—Г–ї—П—А–љ–Њ–є —В–∞—Е–Є–Ї–∞—А–і–Є–Є, —Б—В–µ–њ–µ–љ—М —Д–Є–Ј–Є—З–µ—Б–Ї–Њ–є –љ–∞–≥—А—Г–Ј–Ї–Є –Њ–њ—А–µ–і–µ–ї—П–µ—В —Б–∞–Љ –њ–∞—Ж–Є–µ–љ—В [2]. –Ь—Л —Б—З–Є—В–∞–µ–Љ —Ж–µ–ї–µ—Б–Њ–Њ–±—А–∞–Ј–љ—Л–Љ —А–µ–Ї–Њ–Љ–µ–љ–і–Њ–≤–∞—В—М –њ–Њ–і–≥–Њ—В–Њ–≤–Є—В–µ–ї—М–љ—Г—О¬†–≥—А—Г–њ–њ—Г –њ–Њ¬†—Д–Є–Ј–Ї—Г–ї—М—В—Г—А–µ. –Я—А–Є –љ–∞–ї–Є—З–Є–Є –њ—А–Є–Ј–љ–∞–Ї–Њ–≤ –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є –Ї—А–Њ–≤–Њ–Њ–±—А–∞—Й–µ–љ–Є—П, —Ж–Є–∞–љ–Њ–Ј–∞ —Б–ї–µ–і—Г–µ—В –Є—Б–Ї–ї—О—З–Є—В—М –Ј–∞–љ—П—В–Є—П —Д–Є–Ј–Ї—Г–ї—М—В—Г—А–Њ–є –≤¬†—И–Ї–Њ–ї–µ. –С–Њ–ї–µ–µ —В–Њ—З–љ—Л–Љ –Ї—А–Є—В–µ—А–Є–µ–Љ –Є—Б–Ї–ї—О—З–µ–љ–Є—П –Є–ї–Є —А–∞–Ј—А–µ—И–µ–љ–Є—П —Д–Є–Ј–Є—З–µ—Б–Ї–Є—Е –љ–∞–≥—А—Г–Ј–Њ–Ї –Є¬†–Є—Е —Б—В–µ–њ–µ–љ–Є —П–≤–ї—П—О—В—Б—П —Б—В—А–µ—Б—Б-—В–µ—Б—В—Л. –Т—Л–њ–Њ–ї–љ—П—В—М –Є—Е –Љ–Њ–ґ–љ–Њ —В–Њ–ї—М–Ї–Њ –≤¬†—Б–њ–µ—Ж–Є–∞–ї–Є–Ј–Є—А–Њ–≤–∞–љ–љ—Л—Е —Г—Б–ї–Њ–≤–Є—П—Е.
–Я–Њ—П–≤–ї–µ–љ–Є–µ –њ—А–Є–Ј–љ–∞–Ї–Њ–≤ –і–µ–Ї–Њ–Љ–њ–µ–љ—Б–∞—Ж–Є–Є, —Ж–Є–∞–љ–Њ–Ј–∞ —В—А–µ–±—Г–µ—В –љ–µ–Љ–µ–і–ї–µ–љ–љ–Њ–є –Ї–Њ–љ—Б—Г–ї—М—В–∞—Ж–Є–Є –Ї–∞—А–і–Є–Њ–ї–Њ–≥–∞ –Є¬†–Ї–∞—А–і–Є–Њ—Е–Є—А—Г—А–≥–∞. –•–Є—А—Г—А–≥–Є—З–µ—Б–Ї–Њ–µ –ї–µ—З–µ–љ–Є–µ –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–∞–љ–Њ–Љ–∞–ї–Є–µ–є –≠–±—И—В–µ–є–љ–∞ –Ј–∞–≤–Є—Б–Є—В –Њ—В¬†—Б—В–µ–њ–µ–љ–Є —Б–Љ–µ—Й–µ–љ–Є—П —Б—В–≤–Њ—А–Њ–Ї, –љ–∞—А—Г—И–µ–љ–Є—П —Д—Г–љ–Ї—Ж–Є–є –њ—А–∞–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞¬†[22]. –Э–Њ–≤–Њ—А–Њ–ґ–і–µ–љ–љ—Л–µ —Б¬†—В—П–ґ–µ–ї–Њ–є –∞–љ–Њ–Љ–∞–ї–Є–µ–є –≠–±—И—В–µ–є–љ–∞ –њ–µ—А–µ–і –Њ–њ–µ—А–∞—В–Є–≤–љ—Л–Љ –≤–Љ–µ—И–∞—В–µ–ї—М—Б—В–≤–Њ–Љ –і–ї—П —Б—В–∞–±–Є–ї–Є–Ј–∞—Ж–Є–Є —Б–Њ—Б—В–Њ—П–љ–Є—П –і–Њ–ї–ґ–љ—Л –љ–∞—Е–Њ–і–Є—В—М—Б—П –≤¬†–Њ—В–і–µ–ї–µ–љ–Є–Є –Є–љ—В–µ–љ—Б–Є–≤–љ–Њ–є —В–µ—А–∞–њ–Є–Є. –Э–Њ–≤–Њ—А–Њ–ґ–і–µ–љ–љ—Л–Љ –њ—А–Є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є –ї–µ–≥–Њ—З–љ–Њ–≥–Њ –Ї—А–Њ–≤–Њ—В–Њ–Ї–∞ –Є–ї–Є —Б¬†–і—Г–Ї—В—Г—Б-–Ј–∞–≤–Є—Б–Є–Љ—Л–Љ –Ї—А–Њ–≤–Њ—В–Њ–Ї–Њ–Љ –≤–≤–Њ–і–Є—В—Б—П –њ—А–Њ—Б—В–∞–≥–ї–∞–љ–і–Є–љ –Х1.
–Ч–∞–Ї–ї—О—З–µ–љ–Є–µ
–Р–љ–Њ–Љ–∞–ї–Є—П –≠–±—И—В–µ–є–љ–∞ —Е–∞—А–∞–Ї—В–µ—А–Є–Ј—Г–µ—В—Б—П –∞–њ–Є–Ї–∞–ї—М–љ—Л–Љ —Б–Љ–µ—Й–µ–љ–Є–µ–Љ –њ—А–∞–≤–Њ–≥–Њ –∞—В—А–Є–Њ–≤–µ–љ—В—А–Є–Ї—Г–ї—П—А–љ–Њ–≥–Њ –Ї–Њ–ї—М—Ж–∞ –Є–Ј-–Ј–∞ –љ–∞—А—Г—И–µ–љ–љ–Њ–≥–Њ –Њ—В—Б–ї–Њ–µ–љ–Є—П –Ј–∞–і–љ–µ–є –Є¬†—Б–µ–њ—В–∞–ї—М–љ–Њ–є —Б—В–≤–Њ—А–Њ–Ї –Њ—В¬†–Љ–Є–Њ–Ї–∞—А–і–∞. –І–∞—Б—В–Њ –≤–Њ–Ј–љ–Є–Ї–∞—О—В –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В—М —В—А–Є–Ї—Г—Б–њ–Є–і–∞–ї—М–љ–Њ–≥–Њ –Ї–ї–∞–њ–∞–љ–∞, —А–∞—Б—И–Є—А–µ–љ–Є–µ –њ—А–∞–≤–Њ–≥–Њ –њ—А–µ–і—Б–µ—А–і–Є—П –Є¬†–њ—А–∞–≤–Њ-–ї–µ–≤—Л–є —Б–±—А–Њ—Б —З–µ—А–µ–Ј –і–µ—Д–µ–Ї—В –Љ–µ–ґ–њ—А–µ–і—Б–µ—А–і–љ–Њ–є –њ–µ—А–µ–≥–Њ—А–Њ–і–Ї–Є. –°—А–µ–і–љ–Є–є –≤–Њ–Ј—А–∞—Б—В –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–Є –∞–љ–Њ–Љ–∞–ї–Є–Є –≠–±—И—В–µ–є–љ–∞¬†вАУ 24¬†–≥–Њ–і–∞. –Ю–і–Є–љ –Є–Ј¬†–љ–∞–Є–±–Њ–ї–µ–µ —З–∞—Б—В—Л—Е –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е —Б–Є–Љ–њ—В–Њ–Љ–Њ–≤-–і–µ–±—О—В–∞–љ—В–Њ–≤ (50%)¬†вАУ –∞—А–Є—В–Љ–Є–Є [23].
–Р–љ–Њ–Љ–∞–ї–Є—П –≠–±—И—В–µ–є–љ–∞¬†вАУ —А–µ–і–Ї–Њ–µ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ, —З–∞—Й–µ —Б–њ–Њ—А–∞–і–Є—З–µ—Б–Ї–Њ–µ. –Т¬†–Ї–∞–Ј—Г–Є—Б—В–Є—З–µ—Б–Ї–Є—Е —Б–ї—Г—З–∞—П—Е –Њ–њ–Є—Б—Л–≤–∞—О—В —Б–µ–Љ–µ–є–љ—Л–µ —Д–Њ—А–Љ—Л. –Ь–Є—Б—Б–µ–љ—Б-–Љ—Г—В–∞—Ж–Є—П –Ј–∞–Ї–ї—О—З–∞–µ—В—Б—П (G>A) –≤¬†–Ј–∞–Љ–µ–љ–µ —Н–ї–µ–Ї—В—А–Є—З–µ—Б–Ї–Є –љ–µ–є—В—А–∞–ї—М–љ–Њ–≥–Њ (–љ–µ–њ–Њ–ї—П—А–љ–Њ–≥–Њ)¬†–≥–ї–Є—Ж–Є–љ–∞ –љ–∞¬†–њ–Њ–ї–Њ–ґ–Є—В–µ–ї—М–љ–Њ –Ј–∞—А—П–ґ–µ–љ–љ—Л–є –∞—А–≥–Є–љ–Є–љ. –Э–Њ¬†—Д–µ–љ–Њ—В–Є–њ–Є—З–µ—Б–Ї–Є–є —Б–њ–µ–Ї—В—А —Н—В–Њ–є –Љ—Г—В–∞—Ж–Є–Є –≤¬†FLNA —З—А–µ–Ј–≤—Л—З–∞–є–љ–Њ —И–Є—А–Њ–Ї –Є¬†—Б–≤—П–Ј–∞–љ –њ—А–µ–ґ–і–µ –≤—Б–µ–≥–Њ —Б¬†–∞–љ–Њ–Љ–∞–ї–Є—П–Љ–Є —Б–Ї–µ–ї–µ—В–∞. –£–Ї–∞–Ј–∞–љ–љ–∞—П –Љ—Г—В–∞—Ж–Є—П –Њ—В–Љ–µ—З–µ–љ–∞ –њ—А–Є —Б–Є–љ–і—А–Њ–Љ–∞—Е FG, —Д—А–Њ–љ—В–Њ–Љ–µ—В–∞—Д–Є–Ј–∞–ї—М–љ–Њ–є –і–Є—Б–њ–ї–∞–Ј–Є–Є, —Б–Є–љ–і—А–Њ–Љ–µ –Ь–µ–ї—М–љ–Є–Ї–∞¬†вАУ –Э–Є–і–ї–Ј–∞, –Њ—В–Њ–њ–∞–ї–∞—В–Њ–і–Є–≥–Є—В–∞–ї—М–љ–Њ–Љ —Б–Є–љ–і—А–Њ–Љ–µ —В–Є–њ–Њ–≤ I –Є¬†II. –Ь–Є—Б—Б–µ–љ—Б-–Љ—Г—В–∞—Ж–Є—П –∞—Б—Б–Њ—Ж–Є–Є—А—Г–µ—В—Б—П —В–∞–Ї–ґ–µ —Б¬†—Б–Є–љ–і—А–Њ–Љ–∞–Љ–Є —Б¬†–љ–µ–≤—А–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–Љ–Є (–њ–µ—А–Є–≤–µ–љ—В—А–Є–Ї—Г–ї—П—А–љ–∞—П –љ–Њ–і—Г–ї—П—А–љ–∞—П¬†–≥–µ—В–µ—А–Њ—В–Њ–њ–Є—П) –Є¬†–Ї–∞—А–і–Є–∞–ї—М–љ—Л–Љ–Є (X-—Б–≤—П–Ј–∞–љ–љ–∞—П –і–Є—Б–њ–ї–∞–Ј–Є—П –Ї–ї–∞–њ–∞–љ–Њ–≤) –љ–∞—А—Г—И–µ–љ–Є—П–Љ–Є [24вАУ27]. –Э–Њ¬†–і–ї—П –∞–љ–Њ–Љ–∞–ї–Є–Є –≠–±—И—В–µ–є–љ–∞ —Г–Ї–∞–Ј–∞–љ–љ—Л–µ –љ–∞—А—Г—И–µ–љ–Є—П –љ–µ¬†—Е–∞—А–∞–Ї—В–µ—А–љ—Л, —В–Њ –µ—Б—В—М –Љ–Њ–ґ–љ–Њ –њ—А–µ–і–њ–Њ–ї–Њ–ґ–Є—В—М, —З—В–Њ –Љ—Г—В–∞—Ж–Є—П –≤¬†FLNA¬†вАУ –љ–µ¬†–µ–і–Є–љ—Б—В–≤–µ–љ–љ–∞—П –Є¬†–љ–µ¬†–≥–ї–∞–≤–љ–∞—П –њ—А–Є—З–Є–љ–∞ —Б–њ–Њ—А–∞–і–Є—З–µ—Б–Ї–Њ–є –љ–µ—Б–Є–љ–і—А–Њ–Љ–љ–Њ–є –∞–љ–Њ–Љ–∞–ї–Є–Є –≠–±—И—В–µ–є–љ–∞.
–Ъ–ї–Є–љ–Є—З–µ—Б–Ї–∞—П –Љ–∞–љ–Є—Д–µ—Б—В–∞—Ж–Є—П –њ–Њ—А–Њ–Ї–∞ –Ј–∞–≤–Є—Б–Є—В –Њ—В¬†—Б—В–µ–њ–µ–љ–Є —Б–Љ–µ—Й–µ–љ–Є—П —В—А–Є–Ї—Г—Б–њ–Є–і–∞–ї—М–љ–Њ–≥–Њ –Ї–ї–∞–њ–∞–љ–∞. –Я–∞—Ж–Є–µ–љ—В—Л —Б¬†–њ—А–Є–Ј–љ–∞–Ї–∞–Љ–Є —Б–µ—А–і–µ—З–љ–Њ–є –љ–µ–і–Њ—Б—В–∞—В–Њ—З–љ–Њ—Б—В–Є, —Ж–Є–∞–љ–Њ–Ј–Њ–Љ, —А–∞—Б—И–Є—А–µ–љ–Є–µ–Љ¬†–≥—А–∞–љ–Є—Ж —Б–µ—А–і—Ж–∞ –і–Њ–ї–ґ–љ—Л –±—Л—В—М –љ–µ–Ј–∞–Љ–µ–і–ї–Є—В–µ–ї—М–љ–Њ –љ–∞–њ—А–∞–≤–ї–µ–љ—Л –Ї¬†–Ї–∞—А–і–Є–Њ—Е–Є—А—Г—А–≥—Г.
–†–µ—И–∞—О—Й–Є–Љ –і–ї—П –і–Є–∞–≥–љ–Њ–Ј–∞ –∞–љ–Њ–Љ–∞–ї–Є–Є –≠–±—И—В–µ–є–љ–∞ —П–≤–ї—П–µ—В—Б—П —Г–ї—М—В—А–∞–Ј–≤—Г–Ї–Њ–≤–Њ–µ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–µ. –Т¬†–Њ–њ–Є—Б–∞–љ–Є–Є –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П –і–Њ–ї–ґ–љ—Л –±—Л—В—М –Њ—В—А–∞–ґ–µ–љ—Л –∞–љ–∞—В–Њ–Љ–Є—П —Б—В–≤–Њ—А–Њ–Ї —В—А–Є–Ї—Г—Б–њ–Є–і–∞–ї—М–љ–Њ–≥–Њ –Ї–ї–∞–њ–∞–љ–∞, —Б—В–µ–њ–µ–љ—М —А–µ–≥—Г—А–≥–Є—В–∞—Ж–Є–Є/—Б—В–µ–љ–Њ–Ј–∞, —А–∞–Ј–Љ–µ—А—Л –њ—А–∞–≤–Њ–≥–Њ –њ—А–µ–і—Б–µ—А–і–Є—П –Є¬†–њ—А–∞–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–∞, —Б–≤–µ–і–µ–љ–Є—П –Њ¬†—И—Г–љ—В–Є—А–Њ–≤–∞–љ–Є–Є, —Д—Г–љ–Ї—Ж–Є–Є –Љ–Є–Њ–Ї–∞—А–і–∞ –њ—А–∞–≤–Њ–≥–Њ –Є¬†–ї–µ–≤–Њ–≥–Њ –ґ–µ–ї—Г–і–Њ—З–Ї–Њ–≤, –∞¬†—В–∞–Ї–ґ–µ –≤—Б–µ—Е —Б–Њ–њ—Г—В—Б—В–≤—Г—О—Й–Є—Е –љ–∞—А—Г—И–µ–љ–Є—П—Е.
V.M. Delyagin, PhD, Prof., N.M. Doctorova, I.Ye. Belokrinitskaya, PhD
Dmitry Rogachev National Medical Research Center of Pediatric Hematology, Oncology and Immunology
Scientific and Educational Biomedical Cluster вАШTranslational MedicineвАЩ Peoples' Friendship University of Russia
Contact person: Vasily M. Delyagin, delyagin-doktor@yandex.ru
EbsteinвАЩs anomaly is a rare congenital malformation characterized by apical displacement of the right atrioventricular ring with an anomaly of the development of the tricuspid valve and the frequent presence of other heart defects. According to our data, this defect was diagnosed in 2 cases in more than 6 000 sectional studies: in an adult and a child. According to the echocardiography rooms of non-specialized multidisciplinary children's hospitals and children's polyclinics, the defect is detected with a frequency of 1:24.000вАУ1:32.000 studies. Genetic and environmental factors (viral infections, benzodiazepines, lithium) are indicated as probable causes. Clinical signs are cyanosis, shortness of breath, heart failure, cardiac arrhythmias. Morphologically there are abnormalities of the tricuspid valve, papillary muscles and chordal apparatus, right ventricular myocardial hypertrophy, thinning of the walls of the right atrium and atrialized part of the right ventricle. Decisive for the diagnosis is ultrasound. Displacement of the right atrioventricular ring is revealed. The degree of bias determines the severity of the defect. The amplitude of the tricuspid valve movement is increased. Its valve closure occurs later than the mitral. A 40 ms difference between the tricuspid and mitral valve closure points is a diagnostically important sign of EbsteinвАЩs anomaly. The tactics of a doctor are determined by the clinical picture.
–£–≤–∞–ґ–∞–µ–Љ—Л–є –њ–Њ—Б–µ—В–Є—В–µ–ї—М uMEDp!
–£–≤–µ–і–Њ–Љ–ї—П–µ–Љ –Т–∞—Б –Њ —В–Њ–Љ, —З—В–Њ –Ј–і–µ—Б—М —Б–Њ–і–µ—А–ґ–Є—В—Б—П –Є–љ—Д–Њ—А–Љ–∞—Ж–Є—П, –њ—А–µ–і–љ–∞–Ј–љ–∞—З–µ–љ–љ–∞—П –Є—Б–Ї–ї—О—З–Є—В–µ–ї—М–љ–Њ –і–ї—П —Б–њ–µ—Ж–Є–∞–ї–Є—Б—В–Њ–≤ –Ј–і—А–∞–≤–Њ–Њ—Е—А–∞–љ–µ–љ–Є—П.
–Х—Б–ї–Є –Т—Л –љ–µ —П–≤–ї—П–µ—В–µ—Б—М —Б–њ–µ—Ж–Є–∞–ї–Є—Б—В–Њ–Љ –Ј–і—А–∞–≤–Њ–Њ—Е—А–∞–љ–µ–љ–Є—П, –∞–і–Љ–Є–љ–Є—Б—В—А–∞—Ж–Є—П –љ–µ –љ–µ—Б–µ—В –Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ—Б—В–Є –Ј–∞ –≤–Њ–Ј–Љ–Њ–ґ–љ—Л–µ –Њ—В—А–Є—Ж–∞—В–µ–ї—М–љ—Л–µ –њ–Њ—Б–ї–µ–і—Б—В–≤–Є—П, –≤–Њ–Ј–љ–Є–Ї—И–Є–µ –≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ —Б–∞–Љ–Њ—Б—В–Њ—П—В–µ–ї—М–љ–Њ–≥–Њ –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є—П –Т–∞–Љ–Є –Є–љ—Д–Њ—А–Љ–∞—Ж–Є–Є —Б –њ–Њ—А—В–∞–ї–∞ –±–µ–Ј –њ—А–µ–і–≤–∞—А–Є—В–µ–ї—М–љ–Њ–є –Ї–Њ–љ—Б—Г–ї—М—В–∞—Ж–Є–Є —Б –≤—А–∞—З–Њ–Љ.
–Э–∞–ґ–Є–Љ–∞—П –љ–∞ –Ї–љ–Њ–њ–Ї—Г ¬Ђ–Т–Њ–є—В–Є¬ї, –Т—Л –њ–Њ–і—В–≤–µ—А–ґ–і–∞–µ—В–µ, —З—В–Њ —П–≤–ї—П–µ—В–µ—Б—М –≤—А–∞—З–Њ–Љ –Є–ї–Є —Б—В—Г–і–µ–љ—В–Њ–Љ –Љ–µ–і–Є—Ж–Є–љ—Б–Ї–Њ–≥–Њ –≤—Г–Ј–∞.