Диабетическая нейропатия как мультисистемный процесс
- Аннотация
- Статья
- Ссылки
- English
В статье обсуждаются факторы риска, подробно освещены вопросы патогенеза и новые данные, касающиеся изменения аксонального транспорта нейронов, а также клинического значения этого феномена. Дана характеристика поражения миелинизированных и немиелинизированных нервных волокон. Представлены современные данные о полиорганной вовлеченности в общий патологический процесс при поражении нервной системы, в том числе описаны дисфункция барорефлекса, поражение сетчатки у больных СД, а также ассоциированные с ДПН изменения со стороны центральной нервной системы. Охарактеризованы основные клинические формы диабетических нейропатий. Рассмотрены малоосвещаемые аспекты автономной нейропатии. Сделан акцент на недостаточно внедренных в широкую клиническую практику методах скрининга. Оценены терапевтические возможности, в том числе возможности болезнь-модифицирующей терапии.
В статье обсуждаются факторы риска, подробно освещены вопросы патогенеза и новые данные, касающиеся изменения аксонального транспорта нейронов, а также клинического значения этого феномена. Дана характеристика поражения миелинизированных и немиелинизированных нервных волокон. Представлены современные данные о полиорганной вовлеченности в общий патологический процесс при поражении нервной системы, в том числе описаны дисфункция барорефлекса, поражение сетчатки у больных СД, а также ассоциированные с ДПН изменения со стороны центральной нервной системы. Охарактеризованы основные клинические формы диабетических нейропатий. Рассмотрены малоосвещаемые аспекты автономной нейропатии. Сделан акцент на недостаточно внедренных в широкую клиническую практику методах скрининга. Оценены терапевтические возможности, в том числе возможности болезнь-модифицирующей терапии.
Введение
Отсутствие решения проблемы глобального эпидемического роста заболеваемости сахарным диабетом (СД) не только в краткосрочной, но и в долгосрочной перспективе [1] выводит на первый план вопрос профилактики и лечения его осложнений, которые ложатся дополнительным бременем на систему здравоохранения.
Нервная система (НС) наиболее уязвима при нарушениях углеводного обмена.
Диабетическая полинейропатия (ДПН) является ведущим осложнением СД – встречается более чем у 50% у пациентов с СД [2].
Результаты современных исследований подтверждают все большее значение повреждения периферической НС при СД, ее вклад в развитие ассоциированных с СД патологических состояний.
Процесс начинается рано. У пациентов с предиабетическими нарушениями углеводного обмена полинейропатия отмечается достаточно часто: у 13,00% с нарушенной толерантностью к глюкозе и 11,35% с нарушенной гликемией натощак [3].
Систематический обзор 29 исследований показал широкий диапазон распространенности полинейропатии при предиабете – от 2 до 77% при 95%-ном доверительном интервале (ДИ) 6–34, в большинстве наблюдений она составила ≥ 10% [4].
Клинические проявления и утрата функции НС определяют значительное физическое, психологическое и экономическое бремя полинейропатии [5, 6]. Периферическая нейропатия связана с повышенным риском смерти от всех причин и сердечно-сосудистых заболеваний, особенно у страдающих СД [7].
Диабетические нейропатии представляют собой гетерогенную группу заболеваний, которые можно разделить на генерализованные симметричные полинейропатии (сенсорные, сенсомоторные и вегетативные), а также фокальные и мультифокальные нейропатии, в зависимости от пораженного отдела НС и клинической картины [8].
Среди диабетических нейропатий наиболее частым вариантом является диффузное генерализованное поражение, в частности дистальная симметричная полинейропатия. Она определяется как прогрессирующая потеря аксонов периферических нервов от дистальных отделов к проксимальным, что приводит к снижению чувствительности, возникновению боли и развитию синдрома диабетической стопы [9].
Наиболее распространенные диабетические мононейропатии поражают черепно-мозговые нервы, причем чаще всего поражается третья пара нервов (глазодвигательный). Диабетические радикулопатии локализуются преимущественно в дерматомах груди и живота, никогда не пересекают срединную линию и могут имитировать интраабдоминальные и внутригрудные патологии. Диабетическая амиотрофия связана с поражением поясничного сплетения и представляет собой редкое состояние, проявляющееся сильной болью, мышечной слабостью и атрофией мышц бедра, сопровождающееся выраженной потерей веса.
Однако спектр клинических состояний, ассоциированных с поражением периферической НС при СД, намного шире, чем принято считать в рамках дистальной полинейропатии. Уровень современных знаний о роли изменений НС в развитии ассоциированных с СД патологических состояний требует более детального рассмотрения данного вопроса.
Повреждение периферических нервов может возникать как осложнение по целому спектру неинфекционных метаболических заболеваний, в конечном итоге ассоциированных с диабетом. К ним относятся собственно сам СД, хроническая болезнь почек и, как было недавно установлено, ожирение и метаболический синдром [10, 11]. Хотя каждое из этих состояний имеет уникальную патофизиологию, общей чертой является хроническое системное воздействие измененного гомеостаза. Тонкая структура и функция периферических нервов, особенно чувствительных нейронов, уязвимы для метаболических изменений, вызванных этими системными расстройствами. Саркопения также ассоциирована с метаболическими нарушениями. Данная патология встречается у 10,6% пациентов с СД [12]. Как показал недавно проведенный метаанализ, имеется значительная связь между ДПН и саркопенией. Объединенное отношение шансов (ОШ) составляет 1,62 (95% ДИ 1,30–2,02) [13].
Патофизиология диабетической полинейропатии
Патогенез ДПН – сложное взаимодействие таких факторов, как метаболические процессы, иммунная система, образ жизни и генетическая обусловленность, способствующее повсеместным пагубным модификациям сигнальных путей, что приводит к повреждению нервов [14].
Прогрессирование ДПН включает в себя поражение терминальных сенсорных аксонов на периферии с относительной сохранностью перикарионов (тел клеток).
Модель вовлечения в патологический процесс по типу «носков и перчаток» отражает повреждение сначала самых длинных сенсорных аксонов, например потерю эпидермальных аксонов дистальных отделов ног, предшествующую потере аксонов проксимальных отделов. По этой причине ДПН считается зависимой от роста человека.
Значительное количество экспериментальных данных подтверждают мнение о том, что весь нейрон – от перикариона до терминальных участков аксона служит мишенью для СД [15].
Основными драйверами патогенеза ДПН являются гипергликемия, гиперлипидемия и нарушение передачи сигналов инсулина, что приводит к реализации множества нижележащих метаболических механизмов.
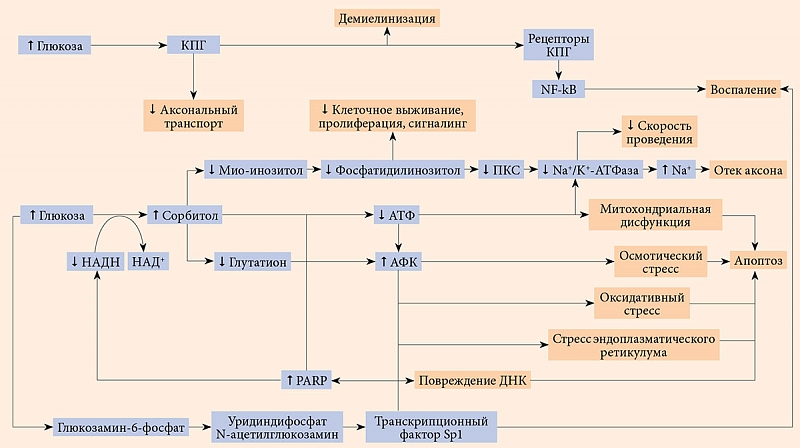
Наиболее изученная патогенная среда – избыточная концентрация глюкозы, которая вызывает гиперактивность полиолового, гликирующего, протеинкиназного С (ПКС), поли(АДФ-рибоза)-полимеразного и гексозаминового путей. Все эти механизмы приводят к развитию оксидативного стресса и одновременному повреждению нервов и микрососудов (рисунок) [16].
Реакция с альдозоредуктазой восстанавливает глюкозу с образованием спиртов, в первую очередь сорбитола, и является первой стадией получения полиолов. Эта реакция требует превращения никотинамидадениндинуклеотида + водорода (Н) (НАДH) в никотинамидадениндинуклеотид (НАД+).
При плохом гликемическом контроле наблюдается чрезмерная активация пути, что приводит к увеличению сорбитола и снижению НАДН. Это связано со снижением мио-инозитола, необходимого для функционирования и развития периферических нервов. При сокращении уровня мио-инозитола преобразуется меньше фосфоинозитида, молекулы, связанной с выживанием клеток, пролиферацией, везикулярной передачей сигналов и метаболизмом глюкозы. В результате изменяется содержание ПКС, которая влияет на натрий-калиевую АТФазу (Na+/K+-АТФазу), фермент, тесно связанный с нервной проводимостью. Накопление иона натрия (Na+) вследствие дисфункции Na+/K+-АТФазы приводит к отеку аксонов. Кроме того, повышенный уровень сорбитола и сниженный уровень НАДН ассоциируются с уменьшением уровня глутатиона, повышением уровня активных форм кислорода (АФК) и клеточной осмолярности. Как следствие, возникают осмотический стресс, стресс эндоплазматического ретикулума и оксидативный стресс, что в сочетании со сниженной продукцией аденозинтрифосфата (АТФ) вызывает повреждение митохондрий и ДНК, снижение кровоснабжения нервов, ускоряя апоптоз. Митоген-активируемая протеинкиназа (mitogen-activated protein kinase – MAPK) – фермент, участвующий как в защитных, так и в апоптотических путях и связанный с альдозоредуктазой и оксидативным стрессом и патогенезом ДПН [17]. Окислительное повреждение вызвано высокой продукцией АФК в отсутствие адекватной антиоксидантной защиты.
В результате избыточного гликирования конечные продукты гликирования (КПГ) могут образовываться в различных клетках (сердца, легких, почек и печени). Развитие ДПН связано с их продукцией в аксонах (как миелинизированных, так и немиелинизированных), перицитах, эндотелиальных и шванновских клетках. КПГ также взаимодействуют с нейрофиламентами и микротрубочками в нервах, что может препятствовать аксональному транспорту, а также приводить к локальной демиелинизации. КПГ ограничивают нейрональный кровоток за счет увеличения проницаемости сосудов, ингибирования вазодилатации за счет интерференции оксида азота (NO), вызывая высвобождение цитокинов и усиление оксидативного стресса. КПГ взаимодействуют с рецепторами КПГ, изменяя сигналинг ядерного фактора каппа B (NF-ĸB), который контролирует несколько воспалительных путей, что приводит к нейродегенерации и утрате способности к регенерации. Кроме того, стимуляция ПКС при гипергликемии связана с сосудистыми модификациями, такими как изменение в структуре эндотелия и внеклеточного матрикса, вазоконстрикция, пролиферация клеток, ангиогенез, активация цитокинов. Интересно, что КПГ стимулируют ПKC в мезангиальных и скелетных мышечных клетках, но эта связь еще не изучена.
Поли(АДФ-рибоза)-полимераза (PARP) опосредует приток свободных радикалов, оксидантов, АФК и активных форм азота в нейроны. В результате взаимодействия NO и супероксида образуется пероксинитрит, который вызывает повреждение ДНК и активирует PARP, фермент репарации ДНК. Чрезмерная активация PARP истощает энергетические ресурсы, о чем свидетельствуют более низкие уровни НАД+ и АТФ, снижает нервную проводимость. Это еще больше усиливает оксидативный и нитрозативный стрессы, повреждение митохондрий, а также апоптоз. По-видимому, PARP также сверхэкспрессирован в микрососудах и может вызывать аналогичный процесс истощения энергии. PARP способен нанести дополнительный ущерб, активируя пути MAPK, гликирования и ПKC, а также влияя на факторы транскрипции и экспрессию генов.
Гипергликемия повышает уровень фруктозо-6-фосфата, таким образом изменяя гексозаминовый путь. Фруктозо-6-фосфат и глутамин реагируют с образованием глюкозамин-6-фосфата, который напрямую может вызывать оксидативный стресс и приводить к повреждению митохондрий и апоптозу. В конечном итоге образуется уридиндифосфат N-ацетилглюкозамин, который в норме служит субстратом для синтеза протеогликанов, однако при ДПН частично неправильно направляется на факторы транскрипции, такие как Sp1. Фактор Sp1 регулирует уровни инсулина и липидов, а также экспрессию трансформирующего фактора роста β1 и ингибитора активатора плазминогена 1 и участвует в развитии воспаления, повреждении ДНК и апоптозе [16, 18].
В контексте СД 1 и 2 типов гипергликемия вносит значительный вклад в повреждение нейронов.
Однако резистентность периферической нейропатии только к строгому гликемическому контролю при СД 2 типа привела к определению роли воспаления и дислипидемии.
Передача сигналов инсулина остается важным фактором с различными эффектами при СД 1 и 2 типов.
Недавно были достигнуты успехи в понимании нейропатии при хронической болезни почек, в связи с чем высказано предположение, что долгосрочная дисрегуляция калия может играть роль в развитии этого состояния.
Строгий контроль уровня глюкозы при СД 2 типа в отличие от такового при СД 1 типа лишь незначительно влияет на исходы периферической нейропатии [19], что ставит под сомнение его возможности модифицировать течение заболевания.
В молекулярных исследованиях выявлено влияние различных липидов на функцию нейронов [20, 21]. Перегрузка субстратом в ответ на дислипидемию при СД способствует активации нескольких путей повреждения клеток. Катаболизм избытка неэстерифицированных жирных кислот приводит к накоплению ацетил-коэнзима А, питающего уже напряженный цикл трикарбоновых кислот, а также к образованию токсичных дезоксисфинголипидов [20]. Накопление липопротеинов низкой плотности активирует каскад передачи сигналов через ряд рецепторов, включая толл-подобный рецептор 4 и рецепторы КПГ, что вызывает образование АФК [22]. Эти провоспалительные сигналы обусловливают воспалительный стресс, стресс эндоплазматического ретикулума и повреждение ДНК. Конвергенция гипергликемических и дислипидемических клеточных путей к оксидативному стрессу и митохондриальному повреждению может представлять собой цель терапевтического вмешательства.
Клетки имеют различные системы защиты для предотвращения или устранения перепроизводства АФК, которые включают антиоксидантные ферменты, такие как супероксиддисмутаза, глутатион и каталаза. Супероксиддисмутаза участвует в удалении супероксидного радикала, способствуя его превращению в H2O2, в то время как глутатион детоксифицирует H2O2 и перекиси липидов [23]. Каталаза ускоряет разложение H2O2 на воду и молекулярный кислород. Избыток глюкозы может нарушать работу антиоксидантной защиты, что влияет на действие ферментов антиоксидантного каскада при СД [24].
Оксидативный стресс является триггером воспаления при СД за счет активации факторов транскрипции (NF-kB), которые влияют на секрецию провоспалительных цитокинов.
Таким образом, в основе развития ДПН лежит повреждение нейрона в результате метаболических, воспалительных и микрососудистых изменений, носящих системный характер.
ДПН обычно характеризуется дисфункцией миелинизированных и немиелинизированных периферических нейронов.
Болевая ДПН связана с дисфункцией преимущественно мелких тонких миелинизированных Аδ-волокон и немиелинизированных С-волокон, тогда как безболевая ДПН – с дисфункцией преимущественно крупных миелинизированных А-волокон. Нередко встречается смешанная потеря мелких и крупных волокон [25].
Микрососудистые изменения могут возникать как при болевой, так и при безболевой ДПН, однако дисфункция микрососудистого кровотока связана с болью [26].
Миелиновая оболочка разрушается на ранних стадиях ДПН.
Так, в образцах икроножного нерва пациентов с минимальными неврологическими проявлениями в начале исследования и через девять лет выявлены межузловые изменения, сегментарная демиелинизация и ремиелинизация. Однако при последующем наблюдении не обнаружена дегенерация аксонов. Это указывает на то, что аксоны становятся мишенями на более позднем этапе заболевания. Немиелинизированные волокна из тех же образцов показали одновременную дегенерацию и регенерацию. Имеющиеся данные свидетельствуют о том, что эти изменения, а также изменения тонких миелинизированных Aδ-волокон происходят до изменений в крупных миелинизированных A-волокнах. R.A. Malik и соавт. представили важную информацию о патогенезе ДПН [27], в частности в отношении функции шванновских клеток. Дальнейшие исследования на культурах шванновских клеток и животных моделях показали, что в гипергликемическом состоянии шванновские клетки продуцируют меньше нейротрофических факторов и подвергаются апоптозу [28, 29]. Эти изменения независимым образом могут способствовать потере аксонов [15, 30].
Сравнение образцов икроножного нерва в группе СД и контрольной группе показало значительные различия в белках цитоскелета.
Нарушение аксонального транспорта и сигналинга – еще один важный патологический элемент при СД, который связан с изменениями в цитоскелете и молекулярных моторных белках, нарушением митохондриального транспорта и нейровоспалением. Аксональный транспорт имеет решающее значение для поддержания гомеостаза нейронов, являясь основным путем связи между телом нейрона и синапсом. Благодаря аксональному транспорту синапс снабжается веществами, необходимыми для нейротрансмиссии и синтезируемыми в теле клетки.
Синаптическую дисфункцию, отмечаемую при СД, связывают и с нарушением аксонального транспорта. Митохондрии также транспортируются по аксонам для обеспечения локальных энергетических потребностей. Кроме того, аксональный транспорт обеспечивает адекватный ответ нейронов на дистальные трофические и стрессовые сигналы.
Таким образом, любой дефект аксонального транспорта может привести к клеточной дисфункции и дегенерации.
При СД в нейронах сетчатки сокращается аксональный транспорт. Нарушение аксонального транспорта является ранним признаком при СД. Оно может способствовать развитию и прогрессированию диабетических осложнений, таких как диабетическая нейропатия, ретинопатия и поражение вещества головного мозга [31].
Диабетическая ретинопатия
Диабетическая ретинопатия – частое осложнение СД. Исторически она считалась микроангиопатическим заболеванием. В настоящее время парадигма смещается в сторону более комплексного взгляда на диабетическую болезнь сетчатки как на тканеспецифическое нервно-сосудистое осложнение, при котором повышенная концентрация глюкозы вызывает не только повреждение микрососудов и ишемию, но и внутриретинальное воспаление и дегенерацию нейронов [32].
Последние данные свидетельствуют о том, что диабетическая ретинопатия является результатом глобальной дисфункции нейроваскулярной единицы. Активация глиальных клеток и дегенерация нейронов признаны событиями патогенеза, которые сочетаются с микрососудистыми изменениями [33, 34]. Внутренняя микроциркуляторная сеть сетчатки лишена вегетативной иннервации, а расширение сосудов обеспечивается глиально-опосредованными ауторегуляторными сигналами от нейронов. В ряде исследований показано, что этот механизм нейроваскулярной связи нарушается уже на ранних стадиях диабетической ретинопатии [35]. Термин «диабетическая болезнь сетчатки» был предложен для обозначения этих патологических нейрососудистых изменений при СД [36].
Связь сахарного диабета с изменениями центральной нервной системы
Связь между СД и изменениями центральной нервной системы (ЦНС) хорошо известна.
СД ассоциируется с 1,5-кратным увеличением риска развития деменции [37].
В нейровизуализационных исследованиях установлено, что у лиц с ДПН могут иметь место дополнительные структурные и функциональные нарушения ЦНС. Кроме того, продемонстрированы различия между пациентами с безболевой и болевой формами ДПН [38].
При ДПН выявлены изменения спинного мозга, ствола мозга, таламуса, соматосенсорной коры.
В 2022 г. S. Smith и соавт. высказали мнение о том, что определение ДПН должно быть пересмотрено с учетом изменений ЦНС и что следует отказаться от общепринятого термина «диабетическая периферическая нейропатия» [16].
Подобные суждения дискутабельны, однако они отражают большой объем новых знаний в области поражения НС при СД.
Диагностика
Диагноз ДПН в первую очередь основывается на клинической оценке, которая включает в себя подробный сбор жалоб, анамнеза, физикальный осмотр, определение неврологического статуса.
Диагностика ДПН не всегда проста и очевидна. Во многих случаях патология протекает асимптомно.
Нужно признать, что существующие диагностические методы для выявления и мониторинга ДПН имеют значительные ограничения, в основном из-за их высокой субъективности, инвазивности и недостаточной повторяемости.
Электронейромиографическое исследование – одно из составляющих оценки ДПН, особенно с учетом вариабельности симптомов и их прогрессирования. Электронейромиография при ДПН стандартизирована и широко применяется в клинической практике, однако она неинформативна при нейропатии тонких волокон.
Золотым стандартом диагностики тонковолоконной нейропатии является биопсия кожи с количественной оценкой плотности внутриэпидермальных нервных волокон, но из-за инвазивности и малодоступности она проводится крайне редко.
Конфокальная микроскопия роговицы – неинвазивный метод офтальмологического осмотра, сравнимый с биопсией кожи при диагностике ДПН [39]. Она позволяет in vivo исследовать все слои роговицы, включая суббазальное нервное сплетение, которое представляет собой часть периферической нервной системы. В частности, от глазной ветви тройничного нерва отходят роговичные нервы, содержащие миелинизированные Aδ-волокна, которые теряют свою миелиновую оболочку в 1 мм от лимба, чтобы гарантировать прозрачность роговицы, и немиелинизированные С-волокна. Данный метод является действенным для обнаружения коррелирующего с тяжестью ДПН повреждения суббазального нервного сплетения [40].
Ранняя диагностика ДПН у лиц с СД имеет большое значение для проведения эффективных целенаправленных мероприятий, позволяющих предотвратить развитие тяжелых осложнений.
Диабетическая вегетативная (автономная) нейропатия
Основные клинические проявления диабетической вегетативной нейропатии включают тахикардию покоя, ортостатическую гипотензию, гастропарез, запор, диарею, недержание кала, эректильную дисфункцию, нейрогенный мочевой пузырь и судомоторную дисфункцию с повышенным или пониженным потоотделением, нераспознавание гипогликемии.
Кардиальная автономная нейропатия (КАН) связана с повышенным риском смерти независимо от наличия других сердечно-сосудистых факторов риска [41].
На ранних стадиях КАН может протекать бессимптомно и обнаруживаться только по снижению вариабельности сердечного ритма при глубоком дыхании. При прогрессировании КАН могут отмечаться тахикардия покоя и ортостатическая гипотензия.
Нарушения автономной регуляции достаточно широко распространены при СД.
Метаанализ, включавший 21 исследование с общим размером выборки 13 772 человека, показал, что распространенность ортостатической гипотензии при СД составляет 24% (95% ДИ 19–28). Среди потенциальных факторов риска указаны повышение уровня гликозилированного гемоглобина (HbA1c) (ОШ 1,13 (95% ДИ 1,07–1,20)), артериальная гипертензия (ОШ 1,02 (95% ДИ 1,01–1,02)) и диабетическая нефропатия (ОШ 2,37 (95% ДИ 1,76–3,19)). Ортостатическая гипотензия в свою очередь связана с более высоким показателем общей смертности и сердечно-сосудистых событий [42].
Барорефлексы позволяют системе кровообращения приспосабливаться к меняющимся условиям жизни при сохранении артериального давления (АД), частоты сердечных сокращений и объема крови в пределах узкого физиологического диапазона.
Барорецепторы встроены в стенки крупных артерий, вен и сердца. Они связаны с ядрами солитарного тракта, расположенного в стволе мозга, через блуждающий и языкоглоточный нервы и активируются при растяжении, когда АД и объем крови увеличиваются. Для противодействия повышению АД барорецепторы вызывают рефлекторное торможение эфферентных симпатических сигналов и расширение сосудов. Сопутствующая парасимпатическая активация синоатриального узла замедляет частоту сердечных сокращений. И наоборот, при переходе в положение стоя барорецепторы разгружаются, что приводит к сужению сосудов и тахикардии с целью компенсировать падение АД. Ортостатическая гипотензия вызвана временным несоответствием между сердечным выбросом и периферическим сосудистым сопротивлением, которое в норме быстро восстанавливается. И артериальные, и сердечно-легочные барорецепторы ингибируют эфферентные симпатические нейроны, приводящие к расширению сосудов, но только артериальные барорецепторы влияют на частоту сердечных сокращений. Артериальные барорецепторы преимущественно нацелены на кровообращение внутренних органов, а сердечно-легочные – ингибируют высвобождение ренина и реабсорбцию натрия в проксимальных канальцах. Активация барорецепторов также подавляет высвобождение вазопрессина [43]. Нарушение барорефлекса предсказывает сердечно-сосудистую смерть и особенно распространено при длительном течении СД.
У пациентов с СД 2 типа дисфункция барорефлекса обнаруживается достаточно рано [44].
Связь между СД 2 типа и сердечной недостаточностью хорошо известна. Однако у пациентов с СД 2 даже при небольшой длительности анамнеза часто может быть выявлено нарушение толерантности к физической нагрузке – маркер, имеющий прогностическое значение. В качестве патофизиологической основы рассматриваются четыре возможных интегрированных механизма, а именно миокардиогенный, миогенный, васкулогенный и нейрогенный. Когда нервный двигательный импульс посылается к скелетной мышце, параллельно импульс передается сердечно-сосудистым центрам в стволе мозга, чтобы увеличить сердечный выброс и АД за счет симпатической активации. Второй детерминантой повышения адренергического симпатического тонуса во время физической нагрузки является мышечный метаборецептор, который стимулируется механической и метаболической активностью мышц. Эти две системы дополнительно модулируются артериальными и сердечно-легочными барорефлексами, которые позволяют оптимально регулировать системные сердечно-сосудистые параметры для удовлетворения региональных метаболических потребностей мышц. Во время упражнений системное периферическое сосудистое русло обычно подвергается симпатической адренергической вазоконстрикции для уменьшения притока крови к тканям, не выполняющим упражнения. В работающих мышцах это уравновешивается локальными сосудорасширяющими механизмами, которые позволяют мышечному кровотоку быстро приспосабливаться к региональным метаболическим потребностям.
При СД 2 типа наблюдается несколько дефектов в нервном контроле описанных выше реакций на физическую нагрузку [45].
Желудочно-кишечные нейропатии могут поражать любую часть желудочно-кишечного тракта. Гастропарез следует заподозрить у лиц с неустойчивым гликемическим контролем или симптомами верхних отделов желудочно-кишечного тракта без другой установленной причины. Исключение органических причин обструкции или язвенной болезни (с помощью эзофагогастродуоденоскопии или визуализации желудка с барием) необходимо до рассмотрения диагноза «гастропарез». Эталонным методом диагностики гастропареза является измерение опорожнения желудка с помощью сцинтиграфии перевариваемых твердых веществ с 15-минутными интервалами в течение 4 часов после приема пищи. Дыхательный тест может использоваться как альтернативный метод диагностики [46].
Острая гипергликемия задерживает эвакуацию из желудка у пациентов с СД.
В исследованиях DCCT и EDIC отсроченная эвакуация была связана с гипергликемией [47].
Влияние гликемического контроля на желудочно-кишечные симптомы при диабетическом гастропарезе малоизучено.
На фоне непрерывной подкожной инфузии инсулина и мониторинга уровня глюкозы оценивались клинические проявления у пациентов с СД и гастропарезом [48]. Через 24 недели у пациентов с исходно плохо контролируемым СД отмечены уменьшение тошноты/рвоты, раннего насыщения и вздутия живота, улучшение качества жизни и увеличение объема пищевых продуктов. Это исследование подтверждает безопасность, осуществимость и потенциальную пользу улучшения гликемического контроля при диабетическом гастропарезе [48].
В то же время установлено, что через шесть месяцев интенсивной терапии, приведшей к снижению уровня HbA1c с 10,6 ± 0,3 до 9,0 ± 0,4%, показатели эвакуации из желудка не изменились [49].
Диабетическая вегетативная невропатия также может обусловливать мочеполовые расстройства, включая сексуальную дисфункцию и дисфункцию мочевого пузыря.
У мужчин диабетическая вегетативная нейропатия способна вызывать эректильную дисфункцию и/или ретроградную эякуляцию. Женская сексуальная дисфункция при СД проявляется снижением полового влечения, усилением боли во время полового акта, снижением полового возбуждения.
Симптомы со стороны нижних мочевыводящих путей представлены недержанием мочи и дисфункцией мочевого пузыря (никтурия, частое мочеиспускание, императивные позывы к мочеиспусканию и слабая струя мочи).
Оценка функции мочевого пузыря должна проводиться у больных СД с рецидивирующими инфекциями мочевыводящих путей, пиелонефритом, недержанием или пальпируемым мочевым пузырем [46].
Нарушение заживления ран – симптом, патогномоничный для СД. В коже находится густая сеть сенсорных нервных афферентов и модуляторов, которые взаимодействуют с эпидермальными кератиноцитами и дермальными фибробластами в двух направлениях, чтобы обеспечить нормальное заживление ран.
Аномальные клеточные реакции, инфекция, иммунологическая и микрососудистая дисфункция и нейропатия вовлечены в патогенез нарушения заживления ран при СД [50].
Дифференциальный диагноз
Диабетическая полинейропатия определяется на основании развития упомянутых выше симптомов и признаков у лиц с СД или предиабетом, при исключении других причин нейропатии [25].
Прежде чем будет установлен диагноз ДПН, необходима дифференциальная диагностика, особенно с учетом того, что существует множество других форм периферической нейропатии, которые могут имитировать ДПН или сочетаться с ней.
Тщательный сбор анамнеза может помочь выявить некоторые из этих состояний, включая злоупотребление алкоголем или другие токсические воздействия, новообразования с историей химиотерапии или амилоидоз.
Кроме того, целесообразен скрининг на гипотиреоз.
Необходимо учитывать также возможную роль почечной недостаточности с уремией.
Дефицит витамина B12 является одним из наиболее распространенных имитаторов ДПН, обнаруживаемых у пациентов с CД 2 типа. Установлено, что до 30% больных СД 2 типа, принимающих метформин, могут иметь дефицит витамина B12 [51]. Таким образом, лабораторный скрининг уровня витамина B12 (в том числе метилмалоновая кислота с гомоцистеином или без него) рекомендуется не реже одного раза в год [2].
Эндокринологам и терапевтам следует помнить и о других причинах, которые диагностируются неврологом: например, воспалительная демиелинизирующая полинейропатия также может имитировать ДПН или сочетаться с ней, подозрение может вызвать внезапное начало и быстрое прогрессирование симптомов.
Необходимо подчеркнуть, что иммуноопосредованные нейропатии являются потенциально опасными для жизни состояниями с уровнем смертности до 13% и 6,6-кратным увеличением смертности от синдрома Гийена – Барре по сравнению с фоновой популяцией того же возраста [52].
Наконец, обнаруживается больше генетических форм полинейропатии и стали доступны новые методы лечения таких заболеваний, как транстиретиновая нейропатия.
Профилактика и лечение
Профилактика
Стратегия профилактики ДПН включает фармакологические и нефармакологические вмешательства.
К нефармакологическим относят модификацию образа жизни, программы реабилитации, например тренировки на выносливость, силу, баланс, сенсомоторные тренировки, обучение и консультирование. Программы упражнений под наблюдением специалиста могут улучшить исходы ДПН [53].
К фармакологическим мерам профилактики относят сахароснижающую терапию. Однако, как известно, усиленный гликемический контроль существенно не снижает риск развития диабетической нейропатии при СД 2 типа [19]. К ограничениям строгого гликемического контроля как метода профилактики ДПН помимо традиционных опасений в отношении эпизодов гипогликемий относят другие побочные эффекты препаратов, а также нейропатию, вызванную лечением (болевая ятрогенная), и, возможно, другие острые нейропатии [54].
Лечение
Терапия ДПН включает в себя модификацию образа жизни, многофакторное вмешательство по поводу СД, а также симптоматическое лечение нейропатической боли и патогенетическое лечение.
Мультимодальное лечение болевого синдрома должно быть направлено не только на облегчение боли, но и на улучшение качества сна, подвижности и общего качества жизни. Полное купирование болевого синдрома при болевой ДПН не всегда возможно. Хорошим результатом считается уменьшение его выраженности на 50% по визуальной аналоговой шкале, а удовлетворительным – на 30%. После уменьшения интенсивности боли обычно достигается основная цель лечения – восстановление или улучшение функционирования пациента и его качества жизни.
Болезнь-модифицирующая терапия. Глубокое понимание патофизиологических механизмов, лежащих в основе ДПН, имеет решающее значение для поиска эффективных методов лечения. Альфа-липоевая кислота (АЛК), бенфотиамин, ацетил-L-карнитин и ряд других препаратов рассматриваются в качестве агентов, направленных на основные патогенетические механизмы ДПН. Из них наиболее значимые результаты получены в отношении АЛК, которая является естественным антиоксидантом, способным уменьшать оксидативный стресс, воздействовать как на невральный, так и на микрососудистый компоненты ДПН.
Основанная на патогенетических механизмах фармакотерапия ориентирована на нейропатический процесс, а не на симптоматическое обезболивание [55].
Получены экспериментальные и клинические данные об эффективности применения препаратов АЛК при поражении нервной системы.
Основанием для суждения об эффективности и безопасности АЛК при ДПН стали результаты двойных слепых рандомизированных клинических исследований ALADIN, ALADIN II, ALADIN III, DEKAN, ORPIL, SYDNEY, SYDNEY II, NATHAN 1 и ряда метаанализов. В частности, в исследовании NATHAN 1 через четыре года было показано, что прием 600 мг АЛК хорошо переносится и уменьшает позитивную неврологическую симптоматику и дефицит [56]. Кроме того, с учетом изложенных выше данных о широком спектре поражений организма при ДПН следует подчеркнуть, что эффекты АЛК доказаны и при автономной нейропатии [57, 58].
Результаты метаанализа свидетельствуют о том, что внутривенное введение АЛК в течение двух – четырех недель безопасно и позволяет значительно улучшить как скорость нервной проводимости, так и нейропатические симптомы [59].
Введение АЛК не только может привести к уменьшению клинических проявлений, но и является безопасным и переносимым вариантом лечения [60].
В настоящее время препараты АЛК широко используются при лечении ДПН.
Установлено, что АЛК ингибирует прогрессирование ДПН и может стимулировать регенерацию нервных волокон путем удаления АФК, регенерации эндогенных и экзогенных антиоксидантов, обновления окисленных белков, ингибирования NF-kB и регуляции транскрипции генов [61].
Необходимо подчеркнуть, что АЛК увеличивает уровень глутатиона, эндогенного антиоксиданта, участвующего в антиоксидантной защите, метаболизме питательных веществ и регуляции клеточных процессов [62]. Глутатион-зависимые ферменты, такие как глутатионпероксидаза, глутатионредуктаза и глутатион-S-трансфераза, являются антиоксидантными ферментами, ответственными за контроль АФК.
С оксидативным стрессом в ЦНС связывают и развитие характерных для СД когнитивных и эмоциональных расстройств.
АЛК повышает чувствительность тканей к инсулину, что может увеличить синтез серотонина и таким образом уменьшить проявления депрессии. В этой связи представляют интерес результаты исследования эффекта перорального приема АЛК в отношении ДПН с оценкой уровня депрессии у 148 пациентов с СД 2 типа [63]. Больные получали комбинированную терапию: гликлазид, ингибитор натрий-глюкозного котранспортера 2, метформин и аналог глюкагоноподобного пептида 1. Пероральный прием 600 мг АЛК через четыре и восемь месяцев привел к статистически значимым результатам. При использовании в сочетании с традиционным лечением значительно снизились частота развития депрессий и проявления периферической нейропатии [63].
Клинические исследования АЛК демонстрируют положительные результаты, в том числе уменьшение боли при ДПН, а также асимметричного диметиларгинина. Кроме того, терапия АЛК ассоциируется с улучшением ряда параметров при оценке ретинопатии [64].
Заключение
Поражение нервной системы при СД носит глобальный характер с вовлечением различных иннервируемых органов и тканей, поэтому не исчерпывается традиционными клиническими характеристиками.
В основе повреждения нейрона лежат системные метаболические, воспалительные и микрососудистые изменения.
Современная медицинская наука позволила существенно расширить представления о патогенетических механизмах ДПН.
Хорошо зарекомендовавшая себя болезнь-модифицирующая терапия ДПН демонстрирует эффекты, актуальные в свете новых знаний о процессах, происходящих в НС при сахарном диабете.
K.V. Antonova, MD, PhD, M.M. Tanashyan, Corresponding member of the RASci., MD, PhD, Prof.
Research Center of Neurology, Moscow
Contact person: Ksenia V. Antonova, kseniya.antonova@mail.ru
Diabetic polyneuropathy (DPN) is the frequent complication of diabetes mellitus (DM) with severe consequences with the progression and defeat of all body systems.
The article discusses risk factors, highlights in detail the issues of pathogenesis and new data concerning changes in axonal transport of neurons, as well as the clinical significance of this phenomenon. Given the characteristic of the lesion of myelinated and non-myelinated nerve fibers. Presented the modern data on multi-organ involvement in the general pathological process in the defeat of the nervous system, including baroreflex dysfunction, retinal damage in patients with DM, as well as changes associated with DPN from the central nervous system. Characterized the main clinical forms of diabetic neuropathies. Considered the little-covered aspects in relation to autonomous neuropathy. The emphasis is placed on screening methods that are insufficiently implemented in the wide clinical practice. Evaluated the therapeutic possibilities, including the possibilities of disease-modifying therapy.
Уважаемый посетитель uMEDp!
Уведомляем Вас о том, что здесь содержится информация, предназначенная исключительно для специалистов здравоохранения.
Если Вы не являетесь специалистом здравоохранения, администрация не несет ответственности за возможные отрицательные последствия, возникшие в результате самостоятельного использования Вами информации с портала без предварительной консультации с врачом.
Нажимая на кнопку «Войти», Вы подтверждаете, что являетесь врачом или студентом медицинского вуза.