Диабетическая полинейропатия: от патогенеза до патогенетической терапии
- Аннотация
- Статья
- Ссылки
- English
В обзоре представлены данные о патофизиологии ДПН с рассмотрением причин последовательности развития симптоматики. Обсуждаются механизмы и значение повреждения митохондрий, нарушения метаболизма и регуляторных функций шванновских клеток, вопросы аутофагии. Отмечена роль таких факторов, как гипергликемия, дислипидемия, инсулинорезистентность и микрососудистые нарушения, описаны сигнальные пути и ассоциированные с ДПН эпигенетические изменения. Подчеркивается роль модификации образа жизни и поведенческих вмешательств в комплексном ведении пациентов с поражением периферической нервной системы при СД. Освещены современные подходы к лечению диабетической нейропатии с точки зрения механизмов, приводящих к ее прогрессированию, а также обозначены нерешенные вопросы и перспективы.
В обзоре представлены данные о патофизиологии ДПН с рассмотрением причин последовательности развития симптоматики. Обсуждаются механизмы и значение повреждения митохондрий, нарушения метаболизма и регуляторных функций шванновских клеток, вопросы аутофагии. Отмечена роль таких факторов, как гипергликемия, дислипидемия, инсулинорезистентность и микрососудистые нарушения, описаны сигнальные пути и ассоциированные с ДПН эпигенетические изменения. Подчеркивается роль модификации образа жизни и поведенческих вмешательств в комплексном ведении пациентов с поражением периферической нервной системы при СД. Освещены современные подходы к лечению диабетической нейропатии с точки зрения механизмов, приводящих к ее прогрессированию, а также обозначены нерешенные вопросы и перспективы.
Введение
Глобальный рост заболеваемости предиабетом и сахарным диабетом (СД) (с предполагаемых 643 млн к 2030 г. до 783 млн к 2045 г. [1]) привел к увеличению распространенности связанных с ними хронических осложнений. Наиболее часто среди таковых встречается диабетическая полинейропатия (ДПН), которая характеризуется широким спектром негативных последствий для здоровья, включая потерю чувствительности, нарушение равновесия, боль, формирование язвы стопы и ампутацию. Лица с ДПН испытывают депрессию и тревогу, у них значительно снижается качество жизни и ограничивается спектр повседневной деятельности. В патологический процесс вовлекается вегетативная нервная система с развитием кардиальной автономной нейропатии и повышенным риском внезапной сердечной смерти [2]. Сопутствующее эпидемии СД увеличение числа случаев ДПН может стать тяжелым бременем как для отдельных лиц, так и для сообществ и иметь медико-социальные и экономические последствия.
Данный обзор призван осветить текущие подходы к лечению диабетической нейропатии, особенно с точки зрения молекулярных механизмов, которые приводят к ее прогрессированию, а также нерешенные вопросы и перспективы изучения заболевания.
Диабетическая нейропатия
Диабетическая нейропатия представляет собой дегенеративное неврологическое заболевание, поражающее соматическую и вегетативную периферическую нервную систему у пациентов с СД в отсутствие какой-либо другой вторичной причины возникновения периферической нейропатии [3].
Термин «диабетическая нейропатия» охватывает широкий спектр нейропатических состояний. На сегодняшний день наиболее распространенным и изученным из них является ДПН. Из всех форм ДПН наибольшую значимость имеет сенсомоторная симметричная (диффузная) дистальная полинейропатия, которая четко коррелирует со снижением качества жизни [4]. Типичная сенсомоторная или преимущественно сенсорная полинейропатия со смешанным паттерном поражения разных типов нервных волокон встречаются наиболее часто – более 90% всех вариантов ДПН. Болевая форма диабетической нейропатии отмечается у 6–34% больных СД [5, 6].
Диабетической нейропатией страдают пациенты с СД 1 и 2 типов. Частота ее встречаемости в течение жизни составляет более 50% [4, 7].
Нейропатия развивается уже на стадии предиабета. Если при предиабете она отмечается в 10–30% случаев, на момент диагностики СД 2 типа – в 20–30% [8]. В дальнейшем эта цифра неуклонно увеличивается [8].
Согласно данным специалистов ФГБНУ «Научный центр неврологии», ДПН отмечалась у 70% лиц с цереброваскулярными заболеваниями на фоне СД 2 типа, что подтверждает глубокую вовлеченность в патологический процесс центральной и периферической нервной системы.
Высокая частота встречаемости диабетической нейропатии в условиях пандемического роста заболеваемости СД определяет острую необходимость комплексных мер по снижению глобального ее бремени для здоровья населения.
Факторы риска развития
Традиционные факторы риска развития ДПН при СД 1 и 2 типов включают плохой гликемический контроль, возраст, длительность диабета и высокий рост [7, 9–11]. С этим согласуются данные нескольких клинических исследований в различных регионах мира, демонстрирующие, что метаболический синдром является фактором риска развития ДПН [7–9, 12]. У лиц с ожирением даже при нормогликемии фиксировалась более высокая распространенность нейропатии по сравнению с лицами без ожирения, а вариабельность уровня глюкозы признана потенциальным фактором риска развития ДПН, особенно болевой формы [13]. Следует подчеркнуть, что заболеваемость ДПН также связана с потенциально модифицируемыми сердечно-сосудистыми факторами риска, включая повышенный уровень триглицеридов, индекс массы тела, курение и артериальную гипертензию [14].
Помимо медицинских важными факторами риска возникновения осложнений СД становятся социальные детерминанты здоровья, наиболее вероятно из-за неадекватного гликемического контроля [15]. Было показано, что у взрослых развитие ДПН связано с низким уровнем образования. Роль общественных факторов подтверждена и в когорте с СД 1 типа. Социальная депривация привела к повышению шансов развития ДПН в 2,17 раза [15]. Болевая форма ДПН ассоциировалась с депрессией, бессонницей и плохим качеством жизни [16].
Патофизиология
При СД наиболее часто поражается периферическая нервная система. ДПН – полинейропатия с поражением длинных нервных волокон, которая развивается в результате метаболических и микрососудистых нарушений на фоне хронической гипергликемии и при наличии факторов сердечно-сосудистого риска. Это заболевание периферической нервной системы, при котором преимущественно поражаются сенсорные и вегетативные аксоны, а позднее в меньшей степени моторные волокна. Такая последовательность событий может быть обусловлена анатомически – аксоны периферической нервной системы имеют длину более 90 см, и они более чем в 20 тыс. раз длиннее поддерживающих их клеточных тел. Сенсорные нейроны периферической нервной системы расположены за пределами гематоэнцефалического барьера, поэтому они более уязвимы к повреждению при гипергликемии по сравнению с мотонейронами, находящимися внутри гематоэнцефалического барьера [16].
Первыми поражаются тонкие немиелинизированные С-волокна, несущие информацию о температурной и болевой чувствительности. В дальнейшем повреждаются миелинизированные волокна – сначала тонкомиелинизированные Aδ-волокна, информирующие о прикосновении, давлении и холоде, затем Aβ- и Aα-волокна, которые отвечают за вибрационную чувствительность и ориентацию тела в пространстве [7].
Патогенез
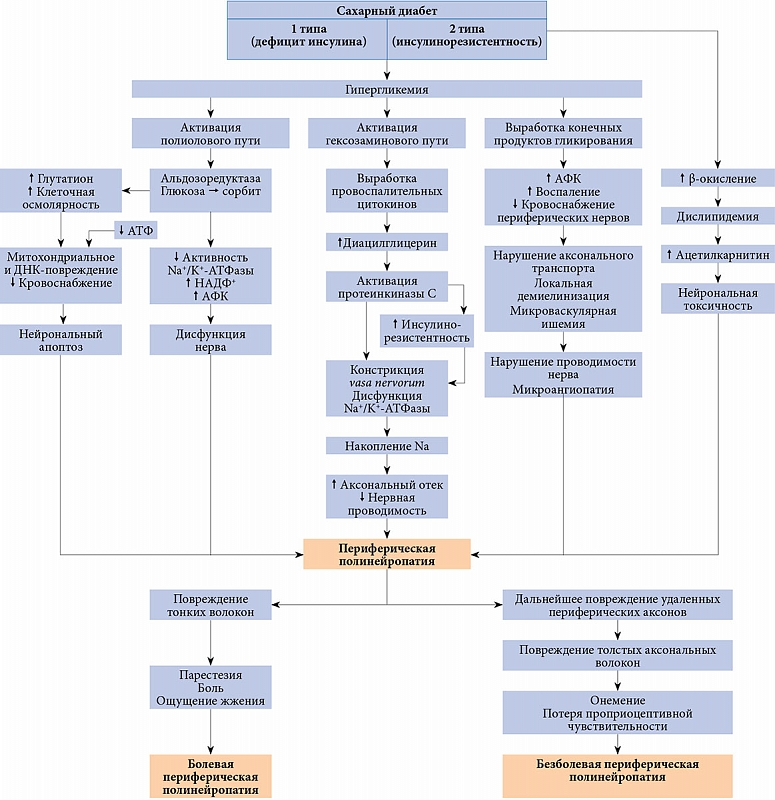
Оксидативный стресс, метаболические нарушения, микроангиопатия и другие факторы, связанные с CД, через специальные пути передачи сигнала разрушают нормальную структуру и функцию нервных клеток и приводят к нейрональной демиелинизации и повреждению нейронов, которые являются основными причинами развития периферической нейропатии [18, 19].
Ранее исследования, направленные на понимание патогенеза диабетической нейропатии, были сосредоточены на оценке роли повышенных концентраций глюкозы [20]. В полиоловом пути альдозоредуктаза превращает избыток глюкозы в сорбит, что в последующем снижает активность натрий-калиевой (Na-K) аденозинтрифосфатазы (АТФазы), истощает запасы никотинамидадениндинуклеотидфосфата (НАДФ) и способствует образованию активных форм кислорода (АФК).
Ингибиторы альдозоредуктазы были протестированы в 32 клинических исследованиях, но достаточный эффект при ДПН не продемонстрирован [7].
Еще одним механизмом может служить то, что избыток глюкозы попадает в гексозаминовый путь, приводя к образованию побочных продуктов воспаления и активации протеинкиназы С. Активация протеинкиназы С усиливает инсулинорезистентность, нарушает активность факторов роста и обусловливает изменение тонуса сосудов.
Необходимо отметить, что исследования ингибиторов протеинкиназы С в качестве терапии ДПН не дали ожидаемого результата [7].
Важный вклад в патогенез могут вносить конечные продукты гликирования (КПГ) и их рецепторы (рКПГ), активация которых приводит к воспалению, накоплению АФК и нарушению кровоснабжения периферических нервов. Накопление КПГ в нейрофиламентах и микротрубочках нервов препятствует аксональному транспорту, в то время как образование КПГ на миелиновой оболочке приводит к локальной демиелинизации. Кроме того, воздействие КПГ на микрососуды увеличивает их проницаемость, препятствует вазодилатации, стимулирует выработку цитокинов и усиливает уровень оксидативного стресса, что в конечном итоге ограничивает кровоснабжение нервов. По мере повреждения большего количества капилляров тесно связанные микрососуды подвергаются ишемии из-за аномальной модификации плотности базальной мембраны, дисфункции перицитов и эндотелиальных клеток и образования артериовенозного шунта [21]. Тяжесть микроангиопатии связана с нарушением проводимости нервов.
Исследования о возможности влияния на КПГ и рКПГ были многообещающими, однако таковое оказалось токсичным для людей [22].
С учетом важнейшей роли гипергликемии в патогенезе ДПН следует подчеркнуть, что интенсивный гликемический контроль не предотвращает ее развития. В настоящее время показано, что риск нейропатии повышается у пациентов с метаболическим синдромом, включающим гипергликемию, ожирение и дислипидемию [7, 23].
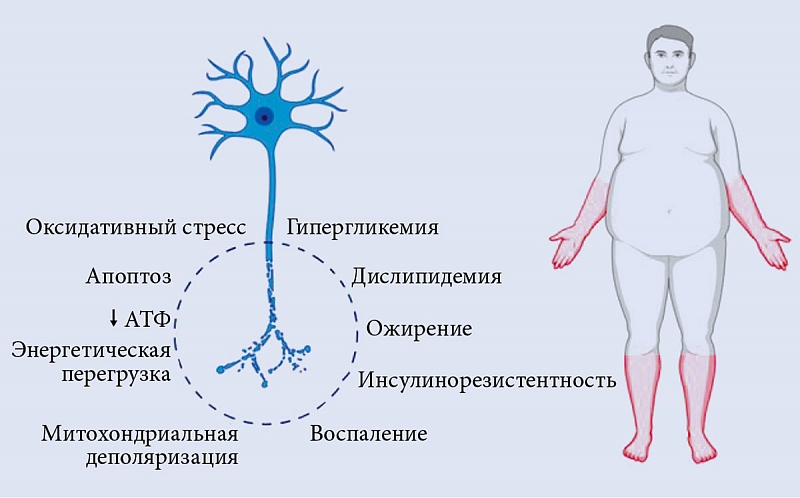
У здоровых лиц инсулин индуцирует высвобождение нейротрофических и нейропротекторных факторов, обеспечивающих выживание нейронов, а также С-пептида, восстанавливает структуру и функцию дефектных аксонов. У лиц с СД 1 типа при снижении уровня инсулина нарушается функция Na+/K+-АТФазы, что приводит к дисфункции нейронов, оксидативному стрессу, отеку аксонов и апоптозу [21]. Инсулинорезистентность при СД 2 типа аналогичным образом может снижать антиоксидантную активность, что способствует митохондриальной дисфункции, перепроизводству оксидативного стресса и апоптозу нейронов [24]. Кроме того, в условиях сопутствующей дислипидемии у пациентов с СД 2 типа свободные жирные кислоты подвергаются активному β-окислению – деградация жирных кислот в митохондриальном матриксе путем окислительного цикла реакций. Трансформация ацетилкофермента А приводит к значительному увеличению содержания ацилкарнитинов, токсичных для нейронов и шванновских клеток [17]. Избыток глюкозы и липидов нарушает нормальные пути их распада и приводит к образованию избыточных доноров электронов, которые не могут обработать клеточные митохондрии. В результате снижается продукция АТФ, нарушается митохондриальный транспорт и накапливаются АФК, что вызывает воспаление, оксидативный стресс и апоптоз нейронов [25]. Оксидативный стресс и митохондриальная дисфункция играют ведущую роль в нейродегенерации [26].
Из-за уменьшения количества функционирующих митохондрий в теле клетки и вдоль аксонов нервные клетки подвергаются дегенерации, при этом наиболее уязвимы удаленные от тела клетки аксоны – в дистальных отделах стопы. Миелинизированные волокна поражаются позже благодаря тому, что шванновские клетки могут обеспечить трофическую функцию и играть протективную роль в условиях повышенных концентраций токсичных веществ. Тонкие волокна не обладают такой защитой, поэтому поражаются первыми [25, 27]. Структура, образованная миелиновой оболочкой и аксоном, имеет радиальную полярность и состоит из различных внутренних мембран, богата рецепторами и молекулами адгезии, которые поддерживают периаксональное пространство и передают сигналы факторов роста [28–30]. Под влиянием гипергликемии и гиперлипидемии, а также через различные сигнальные пути повреждаются митохондрии, нарушаются регуляторные функции шванновских клеток, метаболизм и аутофагия [31–33]. Без защиты и поддержки со стороны глиальных клеток, в частности шванновских клеток, сенсорные нейроны становятся более уязвимыми к повреждению [34, 35].
Индуцированная оксидативным стрессом активация ядерной поли(АДФ-рибоза)-полимеразы 1 (PARP-1) является фундаментальным механизмом развития диабетических осложнений [36, 37]. Это значимый маркер ДПН [38]. PARP-1 – основной подтип PARP, который довольно широко представлен в ядре. PARP-1 играет важную роль в восстановлении ДНК и поддержании целостности генома. На транскрипционном уровне она также регулирует экспрессию белков, таких как медиаторы воспаления, апоптоза и некроза клеток [37]. В исследовании российских пациентов с СД 1 типа установлена связь между геном PARP-1 и патогенезом ДПН [39].
В настоящее время ДПН все чаще рассматривается в аспекте эпигенетических изменений [40]. Низкий уровень метилирования ДНК ассоциирован с развитием диабетической нейропатии у пациентов с СД 2 типа [41]. В недавних исследованиях показано, что СД влияет на гены, имеющие решающее значение для регенерации и функциональности нервов. У больных СД 1 типа с нейропатией наблюдается большее метилирование NINJ2 (ninjurin 2, responsible for nerve regeneration) и более низкое метилирование BRSK2 (BR serine/threonine kinase 2, responsible for nerve functions), чем у пациентов с СД без нейропатии [42]. Снижение копий митохондриальной ДНК также связано с ДПН и гомозиготным вариантом генотипа полиморфизма rs3746444 MIR499A [43]. Генетические вариации MIR-146a, -128a и -27 также повышают предрасположенность к развитию ДПН у пациентов с СД 2 типа [44].
Итак, гипергликемия, дислипидемия, инсулинорезистентность и микрососудистые нарушения являются четырьмя основными факторами, приводящими к развитию ДПН. Гипергликемия и дислипидемия признаны наиболее распространенными факторами, способными запускать путь протеинкиназы С, полиоловый путь, путь КПГ, путь гексозамина и PARP. Изменения инсулинового и других сигнальных путей вызывают изменения периферической нервной системы, такие как воспаление, метаболические нарушения, митохондриальная дисфункция и оксидативный стресс [35].
Механизмы патогенеза диабетической полинейропатии представлены на рис. 1 [45] и обобщены на рис. 2.
Клинические проявления
Как правило, ДПН может длительно протекать бессимптомно. Часто имеет место незаметное нарушение чувствительности и отсутствие боли при очевидной травме. Однако заболевание может дебютировать и с синдрома диабетической стопы [46].
В зависимости от преимущественного поражения толстых или тонких нервных волокон возможны различное начало, течение и клинические проявления [47]. Первыми симптомами чаще всего являются парестезия и дизестезия, которые ощущаются как боль, жжение или покалывание в стопах, что служит признаком дегенерации немиелинизированных тонких С-волокон [7, 48]. По мере прогрессирования ДПН происходит потеря аксонов крупных миелинизированных волокон, при этом пациенты испытывают онемение, вплоть до анестезии стоп, нарушение вибрационной чувствительности и проприоцепции, что приводит к сенсорной атаксии. Обычно нейропатическая боль усиливается в ночное время и значительно ухудшает качество жизни пациентов [48]. Выраженное поражение нервов может приводить к тяжелым осложнениям, таким как синдром диабетической стопы, основными причинами развития которого являются нейропатия и ишемия [7].
Диагностика
На субклинической стадии ДПН определяется только при проведении специальных тестов, с помощью которых оцениваются чувствительные нарушения. Так, может выявляться легкое симметричное снижение болевой чувствительности в дистальных отделах пальцев ног. По мере проксимального прогрессирования формируется типичная картина снижения чувствительности – сначала по типу «носков», позднее по типу «перчаток» [9].
Основным инструментальным методом диагностики ДПН считается стимуляционная электронейромиография, которая также позволяет выявить патологию еще до появления клинических симптомов. С помощью электронейромиографии можно обнаружить дегенеративную аксонопатию – дистальную атрофию и уменьшение крупных и мелких миелинизированных нервных волокон с вторичной дегенерацией, фокальную и сегментарную демиелинизацию. При прогрессировании процесса отмечаются замедление скорости проведения возбуждения в связи с демиелинизацией толстых быстро проводящих волокон, уменьшение амплитуды потенциала действия нервов вследствие аксональной дегенерации [49]. В качестве недостатка данного метода исследования следует отметить возможность оценки функции только толстых миелинизированных нервных волокон Аα- и Аβ-типа. В случае ДПН с преимущественным поражением тонких нервных волокон результат не будет информативным. В такой ситуации следует использовать методики, разработанные специально для диагностики тонковолоконной нейропатии. К ним относятся количественное сенсорное тестирование, биопсия кожи, конфокальная микроскопия роговицы [50].
Ультразвуковое исследование икроножного нерва с измерением площади поперечного сечения может иметь клиническое значение при обследовании больных СД. Будущие исследования в области морфометрии должны быть направлены на выяснение связи с другими характеристиками, такими как параметры тела и возраст. Предельные значения для ДПН, вероятно, должны быть определены для разных географических регионов [51].
Терапия
Ведение пациентов с ДПН должно включать три направления:
- этиотропную терапию, в том числе модификацию образа жизни [7], интенсивную коррекцию гликемии (диабета) [23], коррекцию факторов сердечно-сосудистого риска, в том числе снижение веса при ожирении;
- симптоматическое лечение при наличии нейропатической боли [52];
- патогенетически ориентированную фармакотерапию.
На сегодняшний день получены убедительные доказательства, что поддержание целевой гликемии, близкой к нормальной, предотвращает возникновение и прогрессирование ДПН при СД 1 типа [7, 9]. Однако при СД 1 типа интенсивный гликемический контроль не обеспечивает регресс ДПН. При СД 2 типа эффект контроля уровня глюкозы в отношении ДПН менее убедителен [53]. Вероятными причинами считаются наличие других факторов риска и сопутствующих заболеваний, а также сложных механизмов, приводящих к развитию ДПН при СД 2 типа и описанных выше [2].
Помимо контроля уровня глюкозы многообещающими методами лечения ДПН признаны изменение образа жизни и поведенческие вмешательства. Физические упражнения аналогичны вмешательствам, используемым в программе профилактики развития диабета [54]. Согласно результатам канадского исследования долголетия при СД 1 типа, у пациентов, у которых длительность физической активности составляла 150 минут и более в неделю, частота случаев развития ДПН была на 12% ниже [55]. Физическая активность умеренной интенсивности может сдерживать начало и прогрессирование ДПН у лиц с СД 2 типа, предиабетом и метаболическим синдромом [23]. В контролируемом исследовании мультимодальные аэробные тренировки умеренной интенсивности (50% резерва сердечного ритма) или энергичные упражнения (75% резерва сердечного ритма) улучшали подвижность, равновесие и походку у страдающих ДПН [56]. Кроме того, улучшить симптоматику ДПН способно снижение веса с помощью диеты [57]. Поэтому необходимо приветствовать модификацию образа жизни у больных с установленной полинейропатией.
Систематический обзор и метаанализ рандомизированных контролируемых исследований доказал положительное влияние физических упражнений и на гликемический контроль у больных ДПН. Аэробные тренировки оказались наиболее эффективным способом снижения уровня гликированного гемоглобина (HbA1c), концентрации глюкозы в плазме натощак и после приема пищи.
С метаболической точки зрения диапазон снижения HbA1c имеет клиническое значение для пациентов с полинейропатией [58].
При диабетической нейропатии помимо регулирования уровня глюкозы в крови показаны использование нейротрофических и антиоксидантных средств, ингибирование активности альдозоредуктазы и улучшение микроциркуляции. Патогенетическая терапия направлена не только на облегчение симптомов и боли, но и на основной нейропатический процесс и его клинические последствия.
Некоторые препараты используются в качестве средств, модифицирующих ДПН: альфа-липоевая кислота (АЛК), или тиоктовая кислота, кобаламин. Есть также сообщения о применении эпалрестата и ацетил-L-карнитина [59].
Поскольку оксидативный стресс играет важнейшую роль в патогенезе ДПН, обоснованно использование антиоксидантов [60]. При ДПН наилучшая доказательная база среди антиоксидантов собрана в отношении АЛК [7, 61]. Установлено, что АЛК улучшает симптомы заболевания [62]. Результаты рандомизированных контролируемых исследований и нескольких метаанализов подтвердили, что АЛК эффективна при ДПН [3, 17, 63–66]. АЛК – антиоксидант и кофермент, участвующий в цикле трикарбоновых кислот. Она способна предотвращать и уменьшать риск развития микро- и макрососудистых осложнений, связанных с СД, и улучшать дистальную нервную проводимость. Внутривенные инфузии АЛК в дозе 600 мг/сут снижали выраженность нейропатических симптомов через три недели лечения [60]. При пероральном применении АЛК в дозе 600 мг/сут у пациентов с ДПН клинически значимо уменьшались такие проявления, как боль, парестезия и онемение [67]. В нескольких метаанализах была подтверждена эффективность АЛК при симптоматической ДПН [7, 60]. В исследовании NATHAN 460 пациентов с СД и преимущественно бессимптомной ДПН в течение четырех лет принимали АЛК в дозе 600 мг. Согласно клиническим данным и результатам электронейромиографии, это привело к уменьшению нейропатического дефицита. Был сделан вывод о потенциальной возможности АЛК положительно влиять на патогенетические механизмы ДПН [7, 60]. Результаты исследования NATHAN позволили рассматривать АЛК как препарат для длительного лечения ДПН.
В метаанализе 11 исследований результат оценки по общей шкале неврологических симптомов (Total Symptoms Score, TSS) для АЛК в дозе 600 мг/сут был на 1,05 пункта ниже (стандартная средняя разница -1,05 при 95%-ном доверительном интервале (ДИ) -2,07– -0,04, p = 0,04, I2 = 98,18%), чем для контроля, в конце наблюдения. Согласно данным сетевого метаанализа, прием АЛК в дозе 600 мг позволил достичь значительно более низких значений TSS по сравнению с использованием плацебо (-1,68 (95% ДИ -2,8– -0,6)) [68].
Следует особо отметить позицию относительно наиболее уязвимых категорий больных с ДПН. Нейротропное лечение необходимо проводить на любой стадии процесса, особенно интенсивно и парентерально, у тяжелых больных. Терапия должна включать препараты, блокирующие основные пути повреждения периферических нервов и стимулирующие регенеративные нейропластические процессы [50].
Нерешенные вопросы
История изучения поражения нервной системы при СД началась более полутора тысяч лет назад. К таковому можно отнести описание нейропатической боли у пациента с диабетом и глюкозурией (Susruta (Индия), 500 г.). Однако ни вопросы профилактики и лечения СД, ни проблема развития его осложнений до сих пор не решены.
К актуальным задачам, стоящим перед медицинским сообществом для снижения бремени ДПН, следует отнести:
- разработку персонализированной стратегии профилактики с использованием инновационных технологий и методов искусственного интеллекта;
- изучение клеточных и молекулярных механизмов, с помощью которых изменение образа жизни улучшает нервную функцию;
- поиск механизмов соблюдения стратегий модификации образа жизни;
- разработку алгоритмов эффективных методов лечения, модифицирующих заболевание, особенно болевой ДПН и кардиальной автономной нейропатии;
- разработку и определение информативных чувствительных биомаркеров ДПН;
- поиск новых молекулярных механизмов повреждения и гибели нейронов, связанных с СД (гипергликемия, дислипидемия, инсулинорезистентность);
- изучение взаимодействия между сигнальными путями;
- поиск эффективных и хорошо переносимых методов лечения постуральной гипотензии и гастропареза.
Заключение
Большая распространенность ДПН среди пациентов с СД вызывает тревогу и влечет медико-социальные последствия.
Современные клинические рекомендации сосредоточены на предотвращении прогрессирования и нивелировании симптомов ДПН. Для устранения препятствий к достижению лечебного успеха важно определить механизмы и факторы риска развития ДПН.
Ранняя диагностика позволит своевременно начать патогенетическую терапию с целью профилактики прогрессирования ДПН, а также ее осложнений.
M.M. Tanashyan, Corresponding member of the RASci., MD, PhD, Prof., K.V. Antonova, MD, PhD, N.E. Spryshkov, A.А. Panina, O.V. Lagoda, PhD, E.P. Shchukina, PhD
Federal State Budgetary Scientific Institution ‘Research Center of Neurology’
Federal State Autonomous Educational Institution of Higher Education ‘I.M. Sechenov First Moscow State Medical University’ of the Ministry of Health of the Russian Federation
Contact person: Ksenia V. Antonova, kseniya.antonova@mail.ru
The growing worldwide prevalence of prediabetes and diabetes mellitus (DM) has led to an increase in the incidence of related chronic complications, the most common of which is diabetic polyneuropathy (DPN). The development of DPN is caused by chronic hyperglycemia and potentially modifiable cardiovascular risk factors, including elevated triglyceride levels, body mass index, smoking and hypertension. Social determinants of health are also risk factors for DPN.
The review presents data on the pathophysiology of DPN with consideration of the causes of the sequence of symptoms. Discussed the mechanisms and significance of mitochondrial damage, metabolic disorders and regulatory functions of Schwann cells, and issues of autophagy. Noted the role of factors such as hyperglycemia, dyslipidemia, insulin resistance and microvascular disorders, described the signaling pathways and epigenetic changes associated with DPN. Emphasized the role of lifestyle changes and behavioral interventions in the comprehensive management of patients with lesions of the peripheral nervous system in DM. Highlighted modern approaches to the treatment of diabetic neuropathy from the point of view of the mechanisms leading to its progression, as well as unresolved issues and prospects are outlined.
Уважаемый посетитель uMEDp!
Уведомляем Вас о том, что здесь содержится информация, предназначенная исключительно для специалистов здравоохранения.
Если Вы не являетесь специалистом здравоохранения, администрация не несет ответственности за возможные отрицательные последствия, возникшие в результате самостоятельного использования Вами информации с портала без предварительной консультации с врачом.
Нажимая на кнопку «Войти», Вы подтверждаете, что являетесь врачом или студентом медицинского вуза.