Эффективная и безопасная стартовая терапия сахарного диабета типа 2
- Аннотация
- Статья
- Ссылки




Прошло не одно столетие, прежде чем врачи разделили сахарный диабет на два ведущих типа: тип 1 «инсулинзависимый» и тип 2 «инсулиннезависимый». Наступление «инсулиновой» эры позволило значительно продлить жизнь пациентам, страдающим сахарным диабетом типа 1 (СД тип 1). Но гораздо больше пациентов, страдающих сахарным диабетом типа 2 (СД тип 2). В конце прошлого и начале нынешнего столетия распространенность СД типа 2 приняла масштабы всемирной эпидемии. Так, если в 2000 г. численность больных СД типа 2 в мире составляла около 160 млн человек, то к 2010 году, по прогнозам эпидемиологов, число больных должно было достигнуть 215 млн, а к 2025‑му – более 300 млн человек [1]. Но к началу 2010 года в мире уже было 285 млн заболевших преимущественно СД типа 2. В России всего в 2000 г. оценочно было 8 млн больных СД, а к 2025 г. их количество может увеличиться до 12 млн человек [2, 3].
Патогенез данного заболевания изучен достаточно хорошо. Ведущим фактором в развитии СД типа 2 является инсулинорезистентность (ИР). Антилиполитический эффект инсулина значимо повышается на фоне ИР, что приводит к избыточному накоплению жира, в основном по абдоминальнальному типу. Причем увеличивается количество свободных жирных кислот (СЖК) и глицерина, а, как известно, СЖК являются основным источником формирования атерогенных липопротеинов очень низкой плотности. В печени на фоне инсулинорезистентности повышается процесс глюконеогенеза, и как результат – повышение уровня гликемии натощак. Достоверно установлено, что ИР предшествует клинической манифестации СД типа 2, которая происходит при нарушении функциональной способности более 40% β-клеток. Формирование сосудистых осложнений, которые являются главной причиной инвалидизации пациентов, происходит именно на этом этапе развития заболевания. Пороговыми значениями гликемии, при которых повышается риск развития микро- и макрососудистых осложнений СД типа 2, являются: гликемия натощак > 6,5 ммоль/л, гликемия через 2 часа после еды > 8 ммоль/л, гликированный гемоглобин (HbA1 c) > 7%. Именно поэтому ранняя диагностика и своевременное назначение адекватного лечения являются столь необходимыми. Прежде всего, пациентам рекомендовано изменить привычный образ жизни. На первом этапе необходимо научить пациента правилам рационального питания с исключением из рациона легкоусвояемых углеводов и ограничением употребления жиров. Объяснить необходимость проведения адекватных физических нагрузок и контроля массы тела.
Но только изменение образа жизни не способствует достижению целевых уровней гликемии. Так, по результатам UKPDS 75% пациентов, соблюдающих только принципы изменения образа жизни, к сожалению, в большинстве случаев не достигают целевого уровня HbA1 c < 7%. При недостаточной эффективности изменения образа жизни назначают пероральные сахароснижающие препараты, а в дальнейшем – комбинированную антидиабетическую терапию и инсулинотерапию [3]. Длительное время препаратами «стартовой» терапии считались производные сульфонилмочевины, но в 2005 г. Международная федерация диабета (IDF) рекомендовала метформин в качестве препарата первой линии в сочетании с изменением образа жизни, за исключением пациентов, имеющих патологию почек [4]. Метформин не метаболизируется в организме и экскретируется почками в неизмененном виде. В настоящее время метформин является единственным бигуанидом, рекомендованным для фармакотерапии больных СД типа 2 [23–25]. Основной фармакологический эффект препарата связан со снижением продукции глюкозы печенью, а также продукции свободных жирных кислот (СЖК), окисления жира и, частично, усилением периферического захвата глюкозы [26, 27]. Антигипергликемические эффекты метформина – это результат воздействия препарата на чувствительность к инсулину на уровне печени, мышечной и жировой тканей [28, 29]. Инсулинозависимое поглощение глюкозы, на фоне метформина, повышается на 20–30% [24, 25], происходит подавление процессов глюконеогенеза и в меньшей степени гликогенолиза, что приводит к снижению на 25–30% уровня гликемии натощак [30]. Подавляя клеточное дыхание, метформин ингибирует глюконеогенез и вызывает экспрессию транспортеров глюкозы с последующим улучшением утилизации глюкозы [24, 25, 31]. Метформин противопоказан к применению при нарушении функции почек (снижение клиренса креатинина ниже 50 мл/мин. или повышение креатинина в крови выше 1,5 ммоль/л), печеночной недостаточности, гипоксических состояниях любой этиологии, а также при злоупотреблении алкоголем. Следует воздержаться от назначения препарата в период беременности и лактации, необходима отмена препарата при проведении рентгеноконтрастных исследова-ний в связи с риском развития острой почечной недостаточности и за 5–7 дней до планируемых манипуляций. Метформин следует использовать с осторожностью у пожилых пациентов, со сниженной массой тела. Следует отметить, что риск развития гипогликемии на фоне терапии метформином практически отсутствует, поскольку препарат не стимулирует продукцию инсулина β-клетками [24, 28]. Как бы ни был хорош препарат, но существует часть пациентов с СД типа 2, плохо переносящих метформин в связи с побочными эффектами препарата (диарея, метеоризм, абдоминальный дискомфорт). Вероятно, эти эффекты связаны с накоплением препарата в слизистой кишечника и локальным повышением выработки лактата [23, 37].
Препараты, производные сульфонилмочевины (ПСМ), появились на фармацевтическом рынке таблетированных сахароснижающих средств одними из первых, именно это объясняет столь большую доказательную базу по их эффективности и безопасности. С момента открытия в 1950‑х годах ПСМ являются наиболее широко используемыми при лечении СД типа 2. Ежегодно у 5–10% больных при применении препаратов сульфонилмочевины в виде монотерапии развивается вторичная резистентность [5]. Еще одним частым побочным эффектом ПСМ является развитие гипогликемии. По данным разных авторов, частота ее развития колеблется от 1,8% до 59%, а частота развития тяжелой гипогликемии, требующей оказания пациенту посторонней помощи, составляет 1,9–3,5%. Чаще всего такое состояние возникает у пожилых людей. Иногда тяжелая гипогликемия становится причиной летального исхода в этой группе больных. Вследствие тяжелой гипогликемии могут развиваться судороги, транзиторная гемиплегия, аритмия, инфаркт миокарда и инсульт [6, 7]. Также необходимо учитывать, что риск возникновения гипогликемии увеличивается у пациентов с заболеванием почек, при условии элиминации ПСМ почками (или в случае, если их активные метаболиты выводятся через почки) [8]. Риск развития гипогликемии ограничивает возможность жесткого контроля концентрации глюкозы в крови пациента. Невозможность эффективного гликемического контроля препаратами сульфонилмочевины также можно объяснить прогрессирующим снижением секреции инсулина β-клетками [11], несоблюдением режима питания и объема физических нагрузок, избыточным весом, развитием инфаркта миокарда, инфекционными заболеваниями, а также приемом некоторых препаратов [9]. В проведенных in vitro исследованиях показано, что ПСМ могут стимулировать секрецию инсулина β-клетками поджелудочной железы, независимо от внеклеточной концентрации глюкозы, действуя как секретагоги, частично за счет прямого воздействия на клеточный экзоцитоз [12, 13]. Для оптимального гликемического контроля необходима глюкозозависимая стимуляция β-клеток, которая могла бы обеспечить максимальный контроль постпрандиальной гликемии.
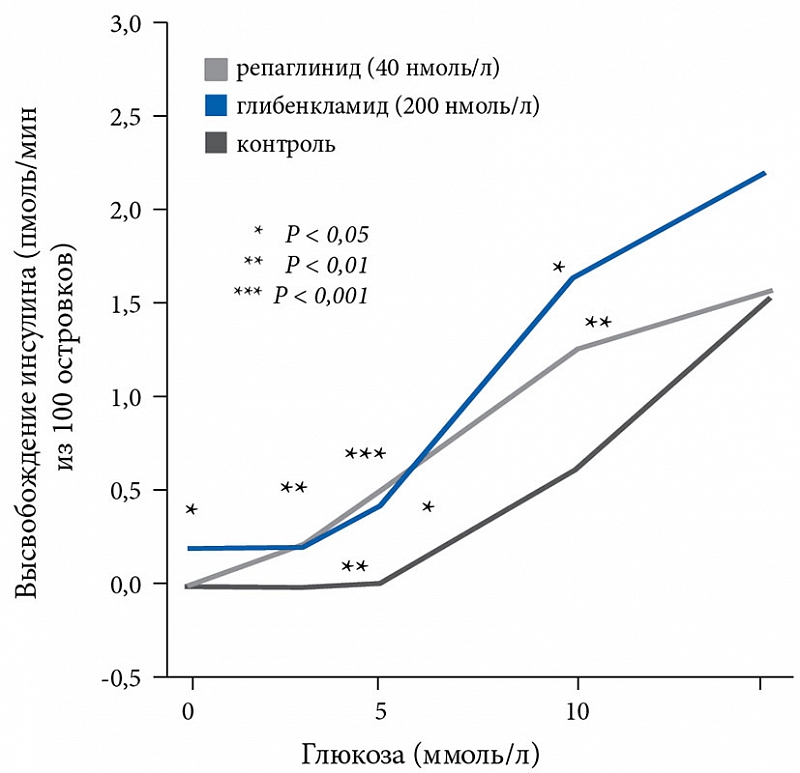
С учетом обозначенной выше необходимости была создана группа меглитинидов, принципиально иной класс химических соединений, имеющий особенные фармакокинетические и фармакодинамические характеристики. Однако секреция инсулина под влиянием репаглинида является лишь частично глюкозозависимой (рис. 1).
Что же можно рекомендовать пациентам с впервые выявленным СД типа 2 при непереносимости метформина?
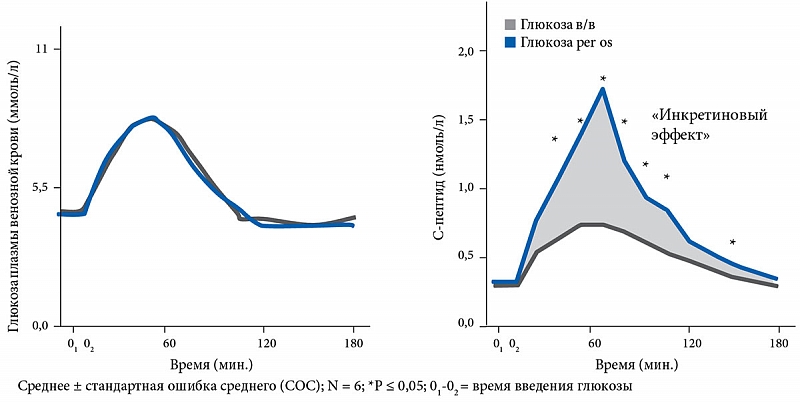
Первые упоминания о неких факторах секретируемых желудочно-кишечным трактом (ЖКТ), высказанных Старлингом, восходят к 1902 г., когда был открыт секретин. В 1930 году Лаббар (Labbarre) ввел термин инкретин, обозначив им гормональную активность кишечника, которая повышает эндокринную секрецию поджелудочной железы. Лишь в 1960 г. стало возможным подтвердить это предположение, когда научились определять уровень инсулина в крови. Был исследован секреторный ответ поджелудочной железы на пероральное и внутривенное введение глюкозы. Доказано, что при одинаковом повышении уровня гликемии секреция инсулина значительно выше при пероральном приеме глюкозы (рис. 2).
Эти результаты свидетельствовали о том, что не только взаимодействие глюкозы с β-клеткой, но и интестинальные факторы участвуют в стимуляции секреции инсулина [38]. Самыми важными гормонами-инкретинами являются: глюкозозависимый инсулинотропный полипептид (ГИП, GIP), прежде известный как желудочный ингибиторный полипептид (ЖИП) и глюкагонподобный пептид –1 (ГПП-1, GLP-1) [39, 40]. У людей без нарушения углеводного обмена до 70% постпрандиальной секреции инсулина обусловлено именно эффектом инкретинов [41, 42], который значительно снижен у больных СД типа 2 и НТГ (рис. 3) [41, 43, 44]. Инсулинотропная активность ГПП-1 четко зависит от уровня гликемии, реализуется путем взаимодействия ГПП-1 со специфическими рецепторами, расположенными на мембране β-клетки [45–47]. Необходимо отметить, что ГПП-1 активирует ген глюкокиназы и ген, кодирующий транспортер глюкозы GLUT 2, которые ответственны за внутриклеточный механизм секреции инсулина [48, 49].
Инфузия ГПП-1 вызывает снижение концентрации глюкозы крови до уровня гликемии натощак [41, 50–51]. Как только уровень гликемии снижается и приближается к нормальным значениям, влияние ГПП-1 на секрецию инсулина прекращается [49]. Клинически важным следствием зависимости эффектов ГПП-1 от уровня глюкозы крови является то, что ГПП-1 не может вызывать гипогликемию. Кроме того, ГПП-1 глюкозозависимым механизмом подавляет секрецию глюкагона панкреатическими β-клетками [48–49]. Наиболее изученным представителем инкретинов является ГПП-1, период его полувыведения составляет 1–2 мин., что делает невозможным применение его в качестве натурального препарата, в клинической практике используются его миметики и аналоги – эксенатид, лираглютид. Инактивация ГПП-1 в организме происходит вследствие отщепления аминокислоты аланина под действием дипептидил пептидазы 4 (ДПП-4). Ингибиторы ДПП-4 продлевают период полужизни инкретинов, благодаря чему усиливается их инсулинотропное действие.
В настоящее время созданы препараты – ингибиторы ДПП-4, защищающие эндогенный ГПП-1 от деградации и удлиняющие его способность снижать постпрандиальную гликемию. Мощным и полностью обратимым ингибитором ДПП-4 является ситаглиптин (Янувия). Благодаря подобному эффекту Янувия способствует повышению в крови уровня интактных инкретинов, влекущих за собой усиление глюкозозависимого инсулинового ответа. Одновременно препарат подавляет секрецию глюкагона. По данным литературы, ситаглиптин обладает протективным влиянием в отношении культуры β-клеток, путем подавления апоптоза и потенциирования пролиферации новых β-клеток [52–54]. Проведены эксперименты на крысах линии Wistar и на мышах с применением стрептозотоцининдуцированного сахарного диабета с нарушением архитектоники островков Лангерганса (распределение β- и α-клеток), с уменьшением числа β-клеток. Назначение ингибиторов DDP-4 позволило улучшить функциональное состояние β-клеток и частично восстановить микроархитектонику островков Лангерганса. В ходе аналогичных экспериментов на мышиной модели с нарушениями секреции и чувствительности к инсулину, при питании богатой жирами пищей лечение ситаглиптином в течение 3 месяцев увеличивало число инсулино-позитивных β-клеток. Кроме того, общая масса β-клеток и отношение числа β-клеток к α-клеткам нормализовалась. При этом восстановилась нормальная архитектура островков [54–59]. У человека, маркерами функции β-клеток служат отношение проинсулин к инсулину и коэффициент HOMA-β, которые значимо улучшаются на фоне терапии препаратом ситаглиптина. Следует отметить, что ситаглиптин достаточно быстро абсорбируется, достигнутая постоянная концентрация в плазме сохраняется в течение 2 дней после однократного приема, метаболизируется в организме, и около 90% принятого препарата экскретируется почками в неизменном виде [60]. По продолжительности действия ситаглиптин превосходит вилдаглиптин [61].
В исследованиях III фазы ситаглиптин изучался в плацебо-контролируемом исследовании, в качестве как монотерапии, так и с добавлением метформина или пиоглитазона. Проведено также клиническое исследование с использованием сульфонилмочевины в качестве препарата сравнения на фоне приема метформина. Рандомизированное двойное слепое плацебо-контролируемое исследование по оценке ОГТТ после однократного приема ситаглиптина. В него были включены 58 пациентов (42 мужчины, 16 женщин; средний возраст 50 лет) с впервые выявленным СД типа 2, HbA1 c 6,5%–11,7% (среднее 8,3%). Произведено распределение по группам: 1‑я группа – пациенты, однократно принявшие ситаглиптин в дозе 25 мг, 2‑я группа – пациенты, однократно принявшие ситаглиптин в дозе 200 мг, и 3‑я контрольная группа, принявшая плацебо. Проведена оценка однократного приема ситаглиптина в двух различных дозах на уровень гликемии (изменение ППК глюкозы) после ОГТТ, а также безопасность и переносимость однократного приема ситаглиптина у пациентов с СД типа 2. Отмечено, что однократный прием ситаглиптина в дозе 200 мг максимально снижал уровень глюкозы после ОГТГ, достоверное улучшение показателей выявлено и при приеме 25 мг препарата по сравнению с плацебо. Основной эффект данной группы заключается в усилении действия ГПП-1 за счет увеличения концентрации данного гормона на фоне снижения инактивации. По данным проведенного исследования, выявлено увеличение концентрации ГПП-1 практически в 2 раза по сравнению с плацебо, причем данный эффект наблюдался в обеих группах и незначительно отличался от дозы принятого препарата. Однократный прием ситаглиптина стимулировал инсулиновый ответ приблизительно на 20% по отношению к плацебо, без активной разницы между концентрацией препарата. Значимая разница между дозами принимаемого препарата была выявлена при оценке ответа при однократном приеме ситаглиптина на подавление секреции глюкагона после ОГТТ. Отмечено, что при приеме 25 мг ситаглиптина секреция глюкагона снижается приблизительно на 7%, а при приеме 200 мг – на 14% по сравнению с плацебо. Оценка клинической безопасности сопоставима с группой плацебо. При однократном приеме препарата тяжелой гипогликемии не наблюдалось.
Необходимо отметить проведенное 24‑недельное плацебо-контролируемое исследование по монотерапии у пациентов с СД типа 2. По дизайну данного протокола пациенты принимали ситаглиптин в дозе 100 мг в сутки или плацебо. Проводился контроль веса, уровня гликемии натощак, гликированного гемоглобина, оценивалось отношение проинсулин/инсулин натощак, рассчитывался индекс HOMA-β, оценивались возможные побочные действия, в том числе и частота развития гипогликемических состояний. Как было написано выше, большинство пациентов, страдающих СД типа 2, имеют избыточную массу тела или ожирение разной степени выраженности. Очень важно, чтобы препарат, способствующий нормализации гликемии, не способствовал увеличению веса, а в идеале приводил к снижению и нормализации ИМТ. При оценке 24‑недельного приема ситаглиптина в дозе 100 мг в сутки не выявлено прибавки массы, отмечено незначительное снижение данного показателя. Подтверждено протективное действие препарата на состояние β-клеток, так как выявлено значимое снижение соотношения проинсулин/инсулин с соответствующим приростом по индексу HOMA-β. Эти показатели достоверно подтверждают способность данного препарата позитивно воздействовать на функцию β-клеток, возможно, на предотвращение апоптоза в них. При анализе показателей гликемии натощак и уровня НвА1с выявлены значимые положительные результаты. Стоит обратить внимание, что в среднем уровень НвА1с снизился на 0,8%, однако интересен факт, что чем выше уровень исходного НвА1с, тем более выражено снижение данного показателя после 24‑недельной терапии ситаглиптином 100 мг.
Но и при «условно хорошем» уровне НвА1с (7%), по сравнению с группой плацебо, более чем у 40% пациентов выявлена положительная динамика. Гипогликемическое состояние бы-ло выявлено у 1,3% пациентов в группе, принимавшей ситаглиптин, и у 0,8% в группе плацебо. Не зафиксировано ни одного тяжелого случая гипогликемии. Из побочных нежелательных явлений достоверно чаще встречались желудочно-кишечные расстройства, которые проявлялись в виде: запоров – 3,8% и диареи – 4,6%, что можно объяснить индивидуальной чувствительностью к данному препарату и что случалось несопоставимо реже, чем аналогичные проявления при приеме препаратов из группы метформина.
Зная основные патогенетические механизмы возникновения и развития СД типа 2 и эффекты действия препаратов ингибиторов ДПП-4 (ситаглипина), можно говорить о необходимости применения данной терапии как патогенетического лечения данного заболевания. Вышеприведенные результаты клинических исследований по эффективности и безопасности применения ситаглиптина позволяют рекомендовать данный препарат как препарат выбора для стартовой терапии у пациентов с СД типа 2 с непереносимостью метформина.
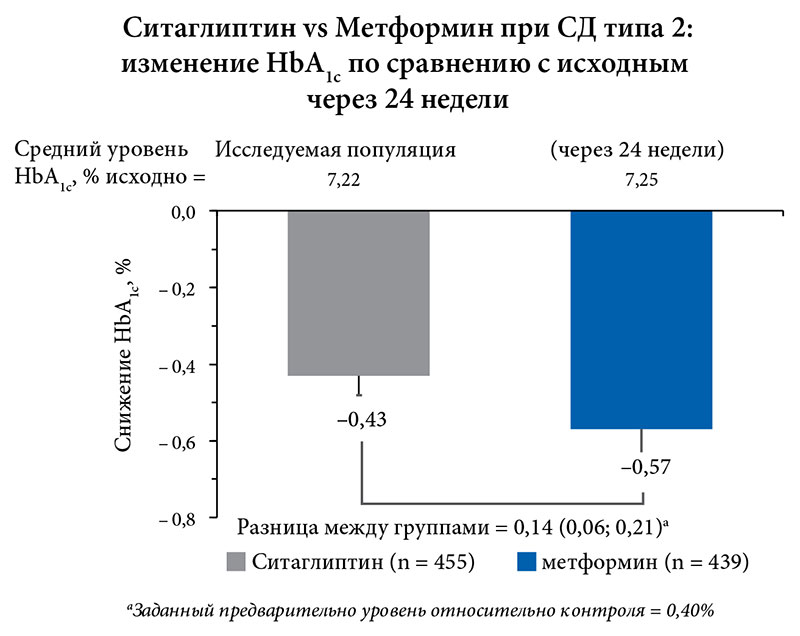
Как отразится на состоянии углеводного обмена подобная стартовая терапия СД типа 2? Объективная невозможность у части пациентов с СД типа 2 начать стартовую терапию по предложенному Консенсусу ADA/EASD без ущерба может быть замещена ситаглиптином. Об этом свидетельствуют результаты 24‑недельного исследования с участием более 1000 пациентов. В этом исследовании проведено сравнение эффективности ситаглиптина и метформина в снижении уровня HbA1c, их эффективность и переносимость у пациентов с СД типа 2 с неадекватным контролем гликемии. Помимо этого, дополнительной целью было сравнение воздействия ситаглиптина и метформина на ЖКТ за 24 недели лечения. Пациенты путем рандомизации были распределены в 2 группы лечения и получали либо ситаглиптин 100 мг 1 раз в сутки, либо метформин 2000 мг/сут. (с постепенной титрацией дозы) (рис. 6).
Результаты исследования показали, что метформин не обладает преимуществом по сравнению с ситаглиптином в нормализации HbA1c (разница между группами не была статистически достоверной): как ситаглиптин, так и метформин вызывали клинически значимое снижение HbA1c (рис. 7).
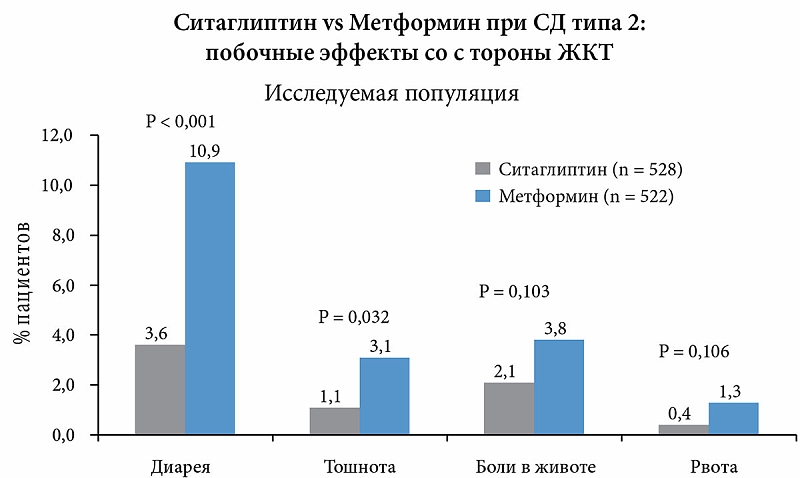
Анализ безопасности и переносимости обоих препаратов показал, что лечение как ситаглиптином, так и метформином в целом переносилось хорошо: частота развития гипогликемии была низкой при применении обоих препаратов; при применении ситаглиптина отмечалась более низкая частота развития НЯ со стороны ЖКТ по сравнению с метформином. Это проявлялось в снижении частоты диареи и тошноты (рис. 8). Лечение как ситаглиптином, так и метформином вызывает некоторое снижение веса тела у больных сахарным диабетом типа 2.
Уважаемый посетитель uMEDp!
Уведомляем Вас о том, что здесь содержится информация, предназначенная исключительно для специалистов здравоохранения.
Если Вы не являетесь специалистом здравоохранения, администрация не несет ответственности за возможные отрицательные последствия, возникшие в результате самостоятельного использования Вами информации с портала без предварительной консультации с врачом.
Нажимая на кнопку «Войти», Вы подтверждаете, что являетесь врачом или студентом медицинского вуза.