Мильгамма® композитум – высокая эффективность при любых формах диабетической нейропатии
- Аннотация
- Статья
- Ссылки
Основные теории, объясняющие развитие осложнений при СД типа 2, включают в себя глюкозотоксичность, инсулинорезистентность / гиперинсулинизм, гипер(дис)липидемию, оксидативный стресс, дисфункцию эндотелия. Сложно сказать, какой механизм первичен, но абсолютно ясно, что они частично запускают друг друга и отягощают течение заболевания, ускоряя процесс формирования диабетических осложнений.
Метаболизм глюкозы у больных СД в результате дефицита инсулина или инсулинорезистентности периферических тканей нарушается. Утилизация глюкозы в инсулинзависимых тканях – печени, жировой ткани, мышцах – осуществляется при участии инсулина. Связываясь со специфическим рецептором на поверхности клеточной мембраны, инсулин способствует экспрессии глюкозных транспортеров (ГЛЮТ 4 в мышечной и жировой ткани) и поступлению глюкозы внутрь клетки. Инсулиннезависимые ткани (эндотелий сосудистой стенки, нервная ткань, хрусталик) также используют глюкозу в качестве энергетического материала, и природа создала все условия для беспрепятственного поступления глюкозы в эти ткани.
Поступление глюкозы в нервную ткань происходит путем пассивной диффузии, без участия инсулина. Для нормального функционирования нервной клетки, помимо глюкозы, необходимо также достаточное поступление кислорода.
В клетке глюкоза расщепляется до пирувата, который в цикле Кребса окисляется с образованием АТФ. Собственные запасы глюкозы и кислорода в нервной клетке крайне малы, и для ее удовлетворительного функционирования необходимо их постоянное поступление в нервные клетки.
В норме глюкоза является самым мощным энергетическим субстратом организма человека, и, казалось бы, чем ее больше, тем лучше, но длительная стойкая гипергликемия приводит к активации комплекса патобиохимических процессов, в результате которых структура белковых субстанций нарушается с образованием конечных продуктов гликирования (КПГ). Данный процесс происходит в организме человека постоянно во всех органах и тканях. Чем длительнее и более выражена гипергликемия, тем выше количество КПГ и гликированных белков. Уровень КПГ коррелирует с процессами старения и степенью выраженности диабетических осложнений, как сосудистых, так и нейропатии.
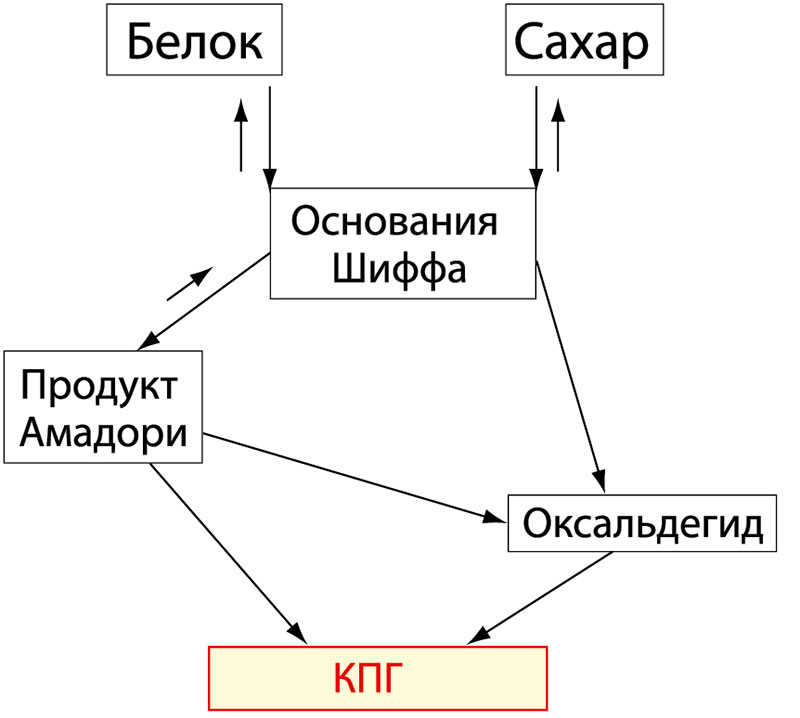
Образование КПГ в организме идет по двум основным путям: один, давно известный «путь Майларда» (рисунок 1), представляет собой взаимодействие молекул сахара и белка с образованием основания Шиффа, которое, являясь нестабильным, преобразуется в продукт Амадори или вновь распадается на молекулу белка и сахара. Продукт Амадори затем преобразуется в КПГ либо прямым превращением, либо через оксальдегид.
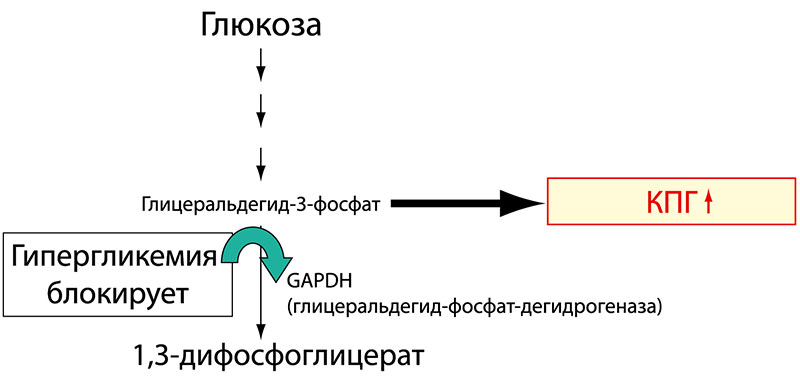
Другой путь образования КПГ стал известен ученым недавно и представляет собой результат метаболизма глюкозы в организме. При гипергликемии продукты метаболизма глюкозы регулируют активность фермента глицеральдегид-фосфат-дегидрогеназы – ключевого фермента, участвующего в регуляции активности всех промежуточных вышележащих метаболитов глюкозы. Снижение активности этого фермента вызывает активацию патобиохимического пути превращения глицеральдегид-3-фосфата в трифосфатоксоальдегид и далее в КПГ (рисунок 2).
Процессы метаболизма глюкозы осуществляются и в теле нейрона, и в его отростках, в шванновских клетках (миелиновой оболочке), следовательно, все отделы нервной ткани способны синтезировать АТФ. Функция нервной клетки заключается в проведении нервного импульса, который зависит от градиента концентрации ионов K+ и Nа+ внутри и вне клетки. АТФ необходима для поддержания активной работы Nа+/K+-АТФазы, фермента, участвующего в генерации нервного импульса и транспорта ионов против градиента концентрации.
Диабетическая нейропатия – наиболее часто встречающееся осложнение СД. По данным некоторых авторов, периферическая форма нейропатии встречается у 95-100% больных. Такие крупные исследования, как UKPDS (1998) и DCCT (1993), доказали зависимость развития осложнений СД от длительности заболевания и компенсации углеводного обмена. Для пациентов СД типа 1 причиной развития является гипергликемия, а частота возникновения нейропатии напрямую коррелируется с длительностью заболевания. Для больных СД типа 2, помимо этого, значимы также гиперлипидемия и системная артериальная гипертония.
Как уже отмечено, главной причиной хронических осложнений СД является гипергликемия. Поддержание нормального уровня глюкозы в крови больных СД является достаточно сложной задачей. Наблюдение за больными СД позволило сделать заключение, что даже очень жесткий контроль уровня гликемии не позволяет избежать развития осложнений. Уровень глюкозы в крови – достаточно мобильный показатель, и избежать значительных колебаний уровня гликемии у больных СД не удается.
Многие поздние осложнения СД являются клиническими проявлениями оксидативного стресса. Косвенным отражением оксидативного стресса является повышение уровня сывороточного оЛПНП, так как повышение антител к оЛПНП положительно коррелирует с выраженностью диабетических осложнений. У пациентов с СД уровень плазменных и сывороточных антиоксидантов значительно снижен.
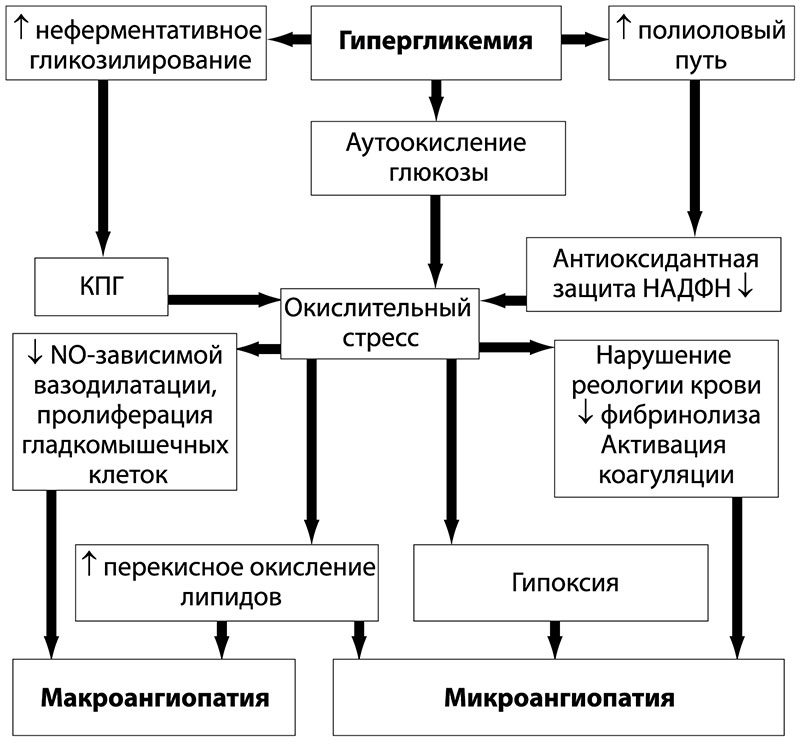
Клеточный оксидативный стресс – это один из важных клеточных элементов, участвующих во многих патологических процессах. Умеренный оксидативный клеточный стресс, кроме того, является общим адаптационным механизмом, лежащим в основе тренирующего эффекта многих факторов физической и химической природы. Окислительный стресс – это один из наиболее важных факторов развития поздних осложнений СД. Условиями для избыточного образования свободных радикалов являются: гипоксия, снижение антиоксидантной защиты, аутоокисление глюкозы, активизация полиолового шунта.
Избыточная продукция кислородсодержащих свободных радикалов способствует окислению мембранных липидов в нервных клетках. «Мишенью» для свободных радикалов является и молекула NО, при окислении которой образуется высокотоксичное вещество ONOO (пироксинитрит). Усиливает деградацию NO повышенное образование супероксидного аниона – продукта окислительного стресса. При активизации полиолового шунта у больных диабетом резко снижается функция эндотелиальной NO-синтетазы – фермента, ответственного за синтез NO из L-аргинина. У здорового человека активность окислительного стресса ограничивается супероксиддисмутазой, каталазой, глутатионпероксидазой, витаминами Е, С, А и восстановленным глутатионом. При гипергликемии снижается активность гликированных ферментов, поэтому превалируют прооксидантные факторы.
Современная фундаментальная наука ключевую роль в инициации повреждения эндоневрального кровотока отводит окислительному стрессу. В экспериментальной эндокринологии доказана эффективность влияния изосорбита нитрата (источник NO) на скорость проведения возбуждения по нервному волокну и улучшение эндоневрального кровотока.
Дефицит системных антиоксидантов усугубляется нарушением всасывания жирорастворимых перехватчиков свободных радикалов вследствие недостаточности экзокринной функции поджелудочной железы. Нарушение всасывания липофильных антиоксидантов усугубляет системный оксидативный стресс, усиливает повреждение органов и тканей свободными радикалами (СР), а также способствует чрезмерному воспалительному ответу. Оксидативный стресс и, как следствие, интенсивное перекисное окисление липидов приводят к изменению мембранного состава клеток иммунной системы и нарушают их активность. СР образуются как промежуточные продукты окисления в дыхательной цепи митохондрий. Один процент всего поглощаемого кислорода утилизируется таким способом и покидает митохондрии в виде частично восстановленных СР, главным образом, в виде О2•-. СР могут повреждать различные ткани, атакуя структурные фосфолипиды, белки, углеводы и ДНК.
Морфологические изменения нервной ткани у больных СД достаточно специфичны и отмечаются во всех отделах центральной, периферической и вегетативной нервной системы. Изменения характеризуются уменьшением числа аксонов в нервных стволах. Вначале поражаются более тонкие, безмиелиновые волокна, в дальнейшем происходит истончение и демиелинизация нервных волокон, повреждение леммоцитов (шванновских клеток) вплоть до полной дегенерации. Все это приводит к денервации тканей, причем дегенеративные изменения происходят и в нервных ганглиях, в результате чего уменьшается число клеточных структур.
В настоящее время клиническая медицина располагает целым арсеналом лекарственных средств как для коррекции гипергликемии – главного виновника сосудистых осложнений, так и лечения этих и других осложнений СД.
Для адекватного терапевтического воздействия необходимо учитывать механизмы, в результате которых происходят подобные изменения. Наиболее доказанными механизмами, влияющими на развитие диабетической нейропатии, являются метаболические и сосудистые. В условиях относительного или абсолютного дефицита инсулина и, как следствие этого – гипергликемии – активизируется полиоловый шунт (рисунок 3). Это путь утилизации глюкозы в инсулиннезависимых тканях, в том числе нервной. В условиях нормогликемии через полиоловый шунт утилизируется всего лишь около 1% глюкозы. При хронической гипергликемии активизируется утилизация глюкозы через полиоловый шунт, вследствие чего происходит частичное истощение ферментных систем (альдозоредуктаза и сорбитолдегидрогеназа). В связи с этим в нервных клетках, леммоцитах происходит накопление сорбитола, а также каскад метаболических нарушений: истощение эндогенного антиоксиданта таурина, усиление аутоокисления глюкозы и накопление кислородсодержащих СР. Накопление сорбитола приводит к осмотическим нарушениям, набуханию клеток и их гибели. Нарушение утилизации глюкозы через полиоловый шунт, снижение образования АТФ и снижение активности Nа+/K+-АТФазы приводит к накоплению внутриклеточного Nа+ и структурным изменениям нейронов, снижению скорости проведения импульса по миелиновым волокнам у больных СД. Накопление сорбитола в клетках способствует снижению поступления миоинозитола в клетку, из которого синтезируется фосфоинозитол – основной регулятор Nа+/K+-АТФазы.
Другим метаболическим нарушением в условиях гипергликемии, как было отмечено выше, является гликирование белков, в том числе нервного волокна. Гликирование – это процесс соединения альдегидной группы углеводной молекулы (глюкозы, фруктозы) и аминогруппы белковой молекулы без участия ферментов. Гликирование миелина приводит к нарушению проводимости по нервному волокну. Гликирование ферментов (альдозредуктазы, сорбитолдегидрогеназы) способствует еще большему энергетическому дефициту нервной клетки.
Наравне с метаболическими нарушениями на развитие нейропатии влияют и изменения эндоневральных капилляров – vasa nervorum. Гипергликемия является причиной развития эндотелиальной дисфункции с избыточной продукцией вазоконстрикторных факторов (эндотелин-1, ангиотензин), подавлением синтеза простациклина, NО (эндотелиального фактора релаксации) – сосудорасширяющих и антиагрегантных факторов. Роль оксида азота (NО) не ограничивается дилатацией локального участка сосудистого русла. Это вещество способно подавлять пролиферативный ответ гладкомышечных клеток сосудистой стенки, блокировать агрегацию тромбоцитов, окисление ЛПНП, адгезию молекул воспаления на эндотелиальных клетках, продукцию эндотелина и др. Изменения мелких сосудов у больных СД характеризуются эндотелиальной деструкцией с отложением фибрина, утолщением и фиброзом медии, избыточной продукцией коллагена, фибронектина, ламенина. Подобные изменения сосудов приводят к развитию ишемической гипоксии нервов, усилению анаэробного гликолиза с избыточным образованием лактата и низким синтезом АТФ.
Первую клиническую классификацию диабетической нейропатии (ДН) предложил В.М. Прихожан (1981), описав центральную и периферическую (ПДН) ее формы, острые и хронические нарушения со стороны центральной и периферической нервной системы. В 1998 г. в Сан-Антонио была принята классификация, подразделяющая ДН на доклиническую и клиническую стадии. Эта классификация ориентирует врачей на возможность профилактических лечебных мероприятий. Клинические проявления диабетической полинейропатии (ДП) возникают, как правило, не ранее чем через 5 лет после манифестации СД типа 1. У пациентов, страдающих СД типа 2, проявления ДН выявляются одновременно с диагностированием заболевания. Явная форма ПДН проявляется у 50-70% больных. Электромиографическое исследование позволяет выявлять ПДН у 100% больных СД. Больные жалуются на мышечную слабость, онемение и боли в ногах, зябкость конечностей, парестезии. Симметрично нарушается температурная, тактильная и болевая чувствительность, развивается гипо- и арефлексия. Проявлением периферических вегетативных поражений является формирование трофических нарушений вплоть до язвенных дефектов тканей, нарушения потоотделения, изменения цвета кожных покровов и др.
Нейропатическая боль причиняет наиболее сильные страдания больным СД и трудно поддается лечению. Большую проблему для врача и пациента составляет диабетическая автономная (висцеральная) нейропатия (ДАН). В 1986 г. D.J. Ewing выделил две группы расстройств вегетативной регуляции у больных СД: клинически явная и бессимптомная ДАН, а также впервые показал зависимость продолжительности жизни больных с ДАН и без нее. Клинические проявления ДАН настолько разнообразны, что зачастую маскируют истинную причину плохого самочувствия больного.
Наиболее опасной формой ДАН является кардиоваскулярная форма. Клиническими проявлениями этой формы являются синусовая тахикардия, ригидный сердечный ритм, ортостатическая гипотония, безболевая форма стенокардии и инфаркта миокарда, характерные изменения ЭКГ – депрессия ST, удлинение интервала QT, инверсия зубца Т. Недостаточная осведомленность врачей, поздняя диагностика и отсутствие терапии в ранние стадии ДАН являются причиной инвалидизации и высокой смертности больных СД.
Диабетическая автономная нейропатия желудочно-кишечного тракта проявляется множественными дисфункциями на всех уровнях: рефлюкс-эзофагиты, дисфагии, тошнота, рвота, атония желудка и гастропарез, энтеропатия с усилением моторики кишечника с профузными поносами или стойкими, не поддающимися терапии запорами. Мочеполовые нарушения характеризуются эректильной дисфункцией у мужчин, мочевым рефлюксом, атонией мочевого пузыря. У больных с длительным течением СД на фоне ДАН нарушается адреналин-опосредованная клиника гипогликемического состояния, которое характеризуется внезапным наступлением гипогликемии и более тяжелым течением.
Основным условием лечения и профилактики осложнений СД является стойкая компенсация углеводного обмена. Такое масштабное исследование, как DCCT, показало, что качество гликемического контроля является основой профилактики и лечения диабетических осложнений. Однако даже тщательный контроль гликемии не всегда препятствует развитию осложнений, и первыми проявлениями осложнений СД являются именно симптомы периферической нейропатии.
Сегодняшний арсенал средств для лечения диабетической нейропатии достаточно широк и имеет патогенетическую направленность. К патогенетической терапии относятся антиоксидантные препараты и метаболические средства (витамины, микроэлементы, ингибиторы альдозоредуктазы, ганглиозиды). Можно сказать, что любая терапия диабетических осложнений сопровождается назначением витаминных препаратов, среди которых основное место занимают витамины группы В, или нейротропные витамины.
Витамин В1 (тиамин). Биологическое значение тиамина обусловлено действием его производного – тиаминдифосфата, образующегося из тиамина и АТФ при участии фермента тиаминкиназы. Тиаминдифосфат является коферментом ряда ферментов, играющих существенную роль в углеводном обмене. Недостаток тиамина в организме ведет к нарушению окисления углеводов, к нарушению зависящих от тиаминдифосфата процессов энергетического и пластического обеспечения жизненных функций, накоплению в крови и тканях недоокисленных продуктов обмена веществ. Участие тиамина в обмене веществ (нервная ткань, сосудистая стенка) определяется коферментной функцией тиаминдифосфата и той ролью, которую в метаболизме играют тиаминдифосфатзависимые ферменты. ТДФ-зависимая пируватдегидрогеназа принимает участие в окислительном декарбоксилировании пировиноградной кислоты с образованием ацетил-КоА и, таким образом, обеспечивается возможность полного окисления углеводов. Другой ТДФ-зависимый фермент – альфакетоглутаратдегидрогеназа участвует в процессах образования янтарной кислоты (сукцината). Этот процесс является важным этапом цикла трикарбоновых кислот. ТДФ принимает участие в процессах окисления кетокислот. ТДФ-зависимая транскетолаза является одним из ферментов пентозофосфатного пути окисления углеводов – пентозного цикла, служащего основным источником НАДНФ-Н и рибозо-5-фосфата. НАДНФ-Н используется в процессах восстановления тканей, в том числе нервной.
В качестве средств симптоматической и патогенетической терапии в медицине применяют витамины группы В1 (тиамин), В6 (пиридоксин) и В12 (цианкобаламин). Это водорастворимые формы витаминов, и при пероральном приеме они являются малоэффективными, так как плохо проникают в ткани организма.
Мильгамма® композитум – золотой стандарт в лечении нейропатии
Мильгамма® композитум содержит уникальное липофильное вещество с тиаминоподобной активностью – бенфотиамин и пиридоксина гидрохлорид. Бенфотиамин в отличие от водорастворимых форм витамина В1 в 10 раз быстрее накапливается в тканях, и почти 100% дозы переходит в активную форму. Достаточное количество бенфотиамина в тканях способствует активности ферментных систем и нейтрализации конечных продуктов гликирования. Оказывается положительное влияние на дегенеративные процессы в нервном волокне, улучшается кровоток в тканях, количество АТФ. Пиридоксин также участвует в метаболизме протеина и частично в метаболизме жиров. Оба витамина потенцируют действие друг друга.
Экспериментальные данные показали влияние бенфотиамина на проявление автономной нейропатии (20). Также в эксперименте изучалось влияние бенфотиамина на развитие ретинопатии, нефропатии и на эндотелиальную функцию. Исследовали крыс со стрептозотоциновым диабетом. Бенфотиамин предотвращал пролиферацию сосудов глазного дна и значительно снижал микроальбуминурию (7). При экспериментальном аллергическом неврите одновременное использование больших доз тиамина, пиридоксина и цианкобаламина показало более позднее развитие неврологической симптоматики и менее выраженную ее форму по сравнению с контролем (8).
Данные клинических плацебо-контролируемых, двойных слепых исследований подтверждают эффективность влияния бенфотиамина и пиридоксина на развитие нейропатий. 20 пациентам с диабетической нейропатией назначали бенфотиамин в дозе 320 мг, пиридоксин и цианкобаламин. Результаты терапии оценивались по шкале неврологических нарушений и вибрационной чувствительности. У пациентов уже через три недели лечения отмечалось достоверное улучшение по этим показателям по сравнению с группой контроля (11).
В другом клиническом исследовании (10) у 24 пациентов с СД 1 и 2 типа проводили лечение препаратом Мильгамма® композитум и об эффективности судили по объективному критерию – скорости проведения нервного импульса. Первые две недели препарат назначался в дозе 320 мг, затем по 120 мг в течение следующих 10 недель. В группе лечения показатели скорости проведения импульса по нерву (n. peroneus) значительно улучшились, и оценка была статистически достоверной. Было также отмечено достоверное улучшение вибрационной чувствительности. Исследование продолжалось в течение одного года, больные получали поддерживающую дозу препарата Мильгамма® композитум. Скорость проведения импульса по нерву и вибрационная чувствительность еще более улучшились.
В плацебо-контролируемом исследовании BEDIP (BEnfotiamine in the treatment of Diabetic Polineuropathy) исследовались 40 пациентов с СД 1 и 2 типа и полинейропатией. В течение 3 недель проводилось лечение бенфотиамином 400 мг в день или плацебо. Показатели нейропатии оценивались по шкале неврологических нарушений и вибрационной чувствительности. Было отмечено достоверное снижение количества баллов по шкале нейропатических нарушений. Достоверных изменений показателей вибрационной чувствительности отмечено не было (12, 13).
Исследовательская группа под руководством профессора А.М. Вейна (1998) отметила улучшение функции вегетативной нервной системы (ВНС) при назначении Мильгаммы® композитум (драже).
Таким образом, терапевтическая эффективность бенфотиамина при диабетической нейропатии может быть патогенетически обоснована. Должно быть обязательным назначение витаминов группы В (Мильгамма®) больным СД в комплексной терапии и в качестве профилактического средства. Для длительной патогенетической и симптоматической терапии желательно использовать препарат, содержащий липофильный бенфотиамин и пиридоксин – Мильгамма® композитум, который обеспечивает усиление и пролонгацию терапевтического эффекта.
Прием Мильгаммы можно рекомендовать и при других заболеваниях, связанных с эндотелиальной дисфункцией, невритах, почечной недостаточности, атеросклерозе.
Уважаемый посетитель uMEDp!
Уведомляем Вас о том, что здесь содержится информация, предназначенная исключительно для специалистов здравоохранения.
Если Вы не являетесь специалистом здравоохранения, администрация не несет ответственности за возможные отрицательные последствия, возникшие в результате самостоятельного использования Вами информации с портала без предварительной консультации с врачом.
Нажимая на кнопку «Войти», Вы подтверждаете, что являетесь врачом или студентом медицинского вуза.