–í–≤–Ķ–ī–Ķ–Ĺ–ł–Ķ
–ď–į—Ā—ā—Ä–ĺ–ł–Ĺ—ā–Ķ—Ā—ā–ł–Ĺ–į–Ľ—Ć–Ĺ—č–Ķ —Ā—ā—Ä–ĺ–ľ–į–Ľ—Ć–Ĺ—č–Ķ –ĺ–Ņ—É—Ö–ĺ–Ľ–ł (gastrointestinal stromal tumours, GIST) —Ź–≤–Ľ—Ź—é—ā—Ā—Ź –Ĺ–į–ł–Ī–ĺ–Ľ–Ķ–Ķ —á–į—Ā—ā—č–ľ–ł –ľ–Ķ–∑–Ķ–Ĺ—Ö–ł–ľ–į–Ľ—Ć–Ĺ—č–ľ–ł –ĺ–Ņ—É—Ö–ĺ–Ľ—Ź–ľ–ł –∂–Ķ–Ľ—É–ī–ĺ—á–Ĺ–ĺ-–ļ–ł—ą–Ķ—á–Ĺ–ĺ–≥–ĺ —ā—Ä–į–ļ—ā–į (–Ė–ö–Ę). –°—á–ł—ā–į–Ķ—ā—Ā—Ź, —á—ā–ĺ GIST –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–ī—Ź—ā –ł–∑ –ļ–Ľ–Ķ—ā–ĺ–ļ –ö–į—Ö–į–Ľ–į (Cajal), –ļ–ĺ—ā–ĺ—Ä—č–Ķ –ĺ—ā–≤–Ķ—á–į—é—ā –∑–į –ľ–ĺ—ā–ĺ—Ä–ł–ļ—É –Ė–ö–Ę. –í –Ņ–ĺ–Ľ—Ć–∑—É —ć—ā–ĺ–≥–ĺ –≥–ĺ–≤–ĺ—Ä–ł—ā –Ĺ–Ķ —ā–ĺ–Ľ—Ć–ļ–ĺ —Ā—Ö–ĺ–∂–Ķ–Ķ –≥–ł—Ā—ā–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ĺ–Ķ —Ā—ā—Ä–ĺ–Ķ–Ĺ–ł–Ķ –Ĺ–į —É–Ľ—Ć—ā—Ä–į—Ā—ā—Ä—É–ļ—ā—É—Ä–Ĺ–ĺ–ľ —É—Ä–ĺ–≤–Ĺ–Ķ, –Ĺ–ĺ –ł —ć–ļ—Ā–Ņ—Ä–Ķ—Ā—Ā–ł—Ź –≥–Ķ–Ĺ–į c-kit –ļ–į–ļ –ļ–Ľ–Ķ—ā–ļ–į–ľ–ł –ö–į—Ö–į–Ľ–į, —ā–į–ļ –ł —Ā—ā—Ä–ĺ–ľ–į–Ľ—Ć–Ĺ—č–ľ–ł –ĺ–Ņ—É—Ö–ĺ–Ľ—Ź–ľ–ł. –≠–ļ—Ā–Ņ—Ä–Ķ—Ā—Ā–ł—Ź –≥–Ķ–Ĺ–į c-kit, –ĺ–Ņ—Ä–Ķ–ī–Ķ–Ľ—Ź–Ķ–ľ–į—Ź —Ā –Ņ–ĺ–ľ–ĺ—Č—Ć—é –ľ–ĺ–Ĺ–ĺ–ļ–Ľ–ĺ–Ĺ–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –į–Ĺ—ā–ł—ā–Ķ–Ľ–į CD117, —Ā—á–ł—ā–į–Ķ—ā—Ā—Ź —Ā—É—Ä—Ä–ĺ–≥–į—ā–Ĺ—č–ľ –ľ–į—Ä–ļ–Ķ—Ä–ĺ–ľ GIST. –í 95% —Ā–Ľ—É—á–į–Ķ–≤ GIST —Ź–≤–Ľ—Ź—é—ā—Ā—Ź –į–Ĺ—ā–ł–≥–Ķ–Ĺ-–Ņ–ĺ–∑–ł—ā–ł–≤–Ĺ—č–ľ–ł –Ņ–ĺ CD117, –ĺ–ī–Ĺ–į–ļ–ĺ –Ņ—Ä–ł –ĺ—ā—Ä–ł—Ü–į—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ļ —Ä–Ķ–į–ļ—Ü–ł–ł –ł –ł—Ā–ļ–Ľ—é—á–Ķ–Ĺ–ł–ł –Ľ–Ķ–Ļ–ĺ–ľ–ł–ĺ—Ā–į—Ä–ļ–ĺ–ľ—č –ľ–ĺ–∂–Ķ—ā –Ņ–ĺ—ā—Ä–Ķ–Ī–ĺ–≤–į—ā—Ć—Ā—Ź –ľ–ĺ–Ľ–Ķ–ļ—É–Ľ—Ź—Ä–Ĺ–ĺ-–≥–Ķ–Ĺ–Ķ—ā–ł—á–Ķ—Ā–ļ–ł–Ļ –į–Ĺ–į–Ľ–ł–∑ —Ā —Ü–Ķ–Ľ—Ć—é –≤—č—Ź–≤–Ľ–Ķ–Ĺ–ł—Ź –į–Ĺ—ā–ł–≥–Ķ–Ĺ-–Ĺ–Ķ–≥–į—ā–ł–≤–Ĺ–ĺ–Ļ –ĺ–Ņ—É—Ö–ĺ–Ľ–ł.¬†
–í¬†2001 –≥. –ī–Ľ—Ź –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź –Ī–ĺ–Ľ—Ć–Ĺ–ĺ–Ļ —Ā –ī–ł—Ā—Ā–Ķ–ľ–ł–Ĺ–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–Ļ —Ą–ĺ—Ä–ľ–ĺ–Ļ GIST –≤–Ņ–Ķ—Ä–≤—č–Ķ –Ī—č–Ľ –Ņ—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā –ď–Ľ–ł–≤–Ķ–ļ (–ł–ľ–į—ā–ł–Ĺ–ł–Ī), –ļ–ĺ—ā–ĺ—Ä—č–Ļ –ĺ–ļ–į–∑–į–Ľ—Ā—Ź —á—Ä–Ķ–∑–≤—č—á–į–Ļ–Ĺ–ĺ —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ—č–ľ –ł –≤—Ā–ļ–ĺ—Ä–Ķ —Ā—ā–į–Ľ —ą–ł—Ä–ĺ–ļ–ĺ –Ņ—Ä–ł–ľ–Ķ–Ĺ—Ź—ā—Ć—Ā—Ź –≤ –ļ–į—á–Ķ—Ā—ā–≤–Ķ –ľ–ĺ–Ĺ–ĺ—ā–Ķ—Ä–į–Ņ–ł–ł –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź, –ļ–ĺ—ā–ĺ—Ä–ĺ–Ķ —Ā—á–ł—ā–į–Ľ–ĺ—Ā—Ć –Ĺ–Ķ—á—É–≤—Ā—ā–≤–ł—ā–Ķ–Ľ—Ć–Ĺ—č–ľ –ļ —Ö–ł–ľ–ł–ĺ–Ľ—É—á–Ķ–≤–ĺ–ľ—É –Ľ–Ķ—á–Ķ–Ĺ–ł—é. –ü—Ä–į–ļ—ā–ł—á–Ķ—Ā–ļ–ł —Ā—Ä–į–∑—É –Ĺ–į—á–į–Ľ–ł –Ņ—Ä–ĺ–≤–ĺ–ī–ł—ā—Ć—Ā—Ź –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź –Ņ–ĺ –ł–∑—É—á–Ķ–Ĺ–ł—é —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł –ď–Ľ–ł–≤–Ķ–ļ–į –≤ –į–ī—ä—é–≤–į–Ĺ—ā–Ĺ–ĺ–ľ –ł –Ĺ–Ķ–ĺ–į–ī—ä—é–≤–į–Ĺ—ā–Ĺ–ĺ–ľ —Ä–Ķ–∂–ł–ľ–į—Ö. –í 2011 –≥. –Ĺ–į —Ā—ä–Ķ–∑–ī–Ķ –ź–ľ–Ķ—Ä–ł–ļ–į–Ĺ—Ā–ļ–ĺ–≥–ĺ –ĺ–Ī—Č–Ķ—Ā—ā–≤–į –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ļ –ĺ–Ĺ–ļ–ĺ–Ľ–ĺ–≥–ł–ł (American Society of Clinical Oncology, ASCO) –Ī—č–Ľ–ĺ —Ā–ĺ–ĺ–Ī—Č–Ķ–Ĺ–ĺ –ĺ–Ī —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł 3-–Ľ–Ķ—ā–Ĺ–Ķ–Ļ –į–ī—ä—é–≤–į–Ĺ—ā–Ĺ–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł –ď–Ľ–ł–≤–Ķ–ļ–ĺ–ľ —É –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö —Ā –≤—č—Ā–ĺ–ļ–ł–ľ —Ä–ł—Ā–ļ–ĺ–ľ –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź. –ú–Ĺ–ĺ–≥–ĺ—á–ł—Ā–Ľ–Ķ–Ĺ–Ĺ—č–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź —ā–į–ļ–∂–Ķ –Ņ–ĺ–ļ–į–∑–į–Ľ–ł, —á—ā–ĺ –ĺ—Ā–Ĺ–ĺ–≤–Ĺ—č–Ķ –≥–ł—Ā—ā–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł–Ķ —Ö–į—Ä–į–ļ—ā–Ķ—Ä–ł—Ā—ā–ł–ļ–ł (—Ä–į–∑–ľ–Ķ—Ä –ĺ–Ņ—É—Ö–ĺ–Ľ–ł, –Ľ–ĺ–ļ–į–Ľ–ł–∑–į—Ü–ł—Ź, –ľ–ł—ā–ĺ—ā–ł—á–Ķ—Ā–ļ–ł–Ļ –ł–Ĺ–ī–Ķ–ļ—Ā), –į —ā–į–ļ–∂–Ķ –ľ–ĺ–Ľ–Ķ–ļ—É–Ľ—Ź—Ä–Ĺ–ĺ-–≥–Ķ–Ĺ–Ķ—ā–ł—á–Ķ—Ā–ļ–ł–Ļ —Ā—ā–į—ā—É—Ā —Ź–≤–Ľ—Ź—é—ā—Ā—Ź –≤–į–∂–Ĺ—č–ľ–ł —Ą–į–ļ—ā–ĺ—Ä–į–ľ–ł –Ņ—Ä–ĺ–≥–Ĺ–ĺ–∑–į.¬†
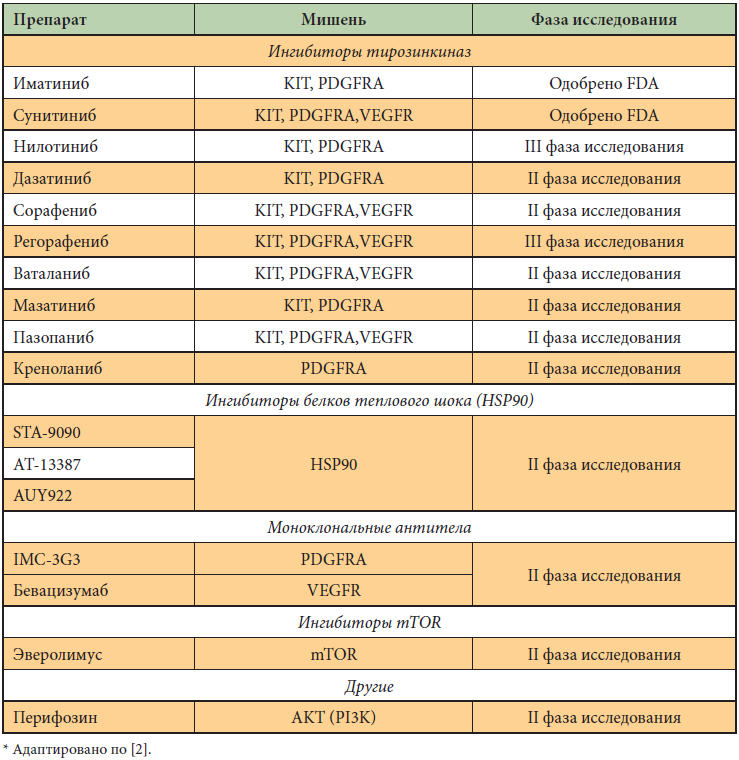
–Ę–į–ļ, –Ĺ–į–Ņ—Ä–ł–ľ–Ķ—Ä, –Ņ–į—Ü–ł–Ķ–Ĺ—ā—č —Ā –ľ–Ķ—ā–į—Ā—ā–į—ā–ł—á–Ķ—Ā–ļ–ĺ–Ļ —Ą–ĺ—Ä–ľ–ĺ–Ļ –Ī–ĺ–Ľ–Ķ–∑–Ĺ–ł –ł –ľ—É—ā–į—Ü–ł–Ķ–Ļ –≤ 9-–ľ —ć–ļ–∑–ĺ–Ĺ–Ķ –≥–Ķ–Ĺ–į c-kit –Ņ–ĺ–Ľ—É—á–į—é—ā –≤—č–≥–ĺ–ī—É –≤ –Ņ–Ľ–į–Ĺ–Ķ –Ī–Ķ–∑—Ä–Ķ—Ü–ł–ī–ł–≤–Ĺ–ĺ–Ļ –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā–ł, –Ķ—Ā–Ľ–ł —Ā—Ä–į–∑—É –Ņ–ĺ–Ľ—É—á–į—é—ā –ī–≤–ĺ–Ļ–Ĺ—É—é –ī–ĺ–∑—É –Ľ–Ķ–ļ–į—Ä—Ā—ā–≤–į. –ü–į—Ü–ł–Ķ–Ĺ—ā—č —Ā –ļ—Ä–į–Ļ–Ĺ–Ķ —Ä–Ķ–ī–ļ–ĺ–Ļ –ľ—É—ā–į—Ü–ł–Ķ–Ļ D842V –Ņ–ĺ–Ľ–Ĺ–ĺ—Ā—ā—Ć—é –Ĺ–Ķ—á—É–≤—Ā—ā–≤–ł—ā–Ķ–Ľ—Ć–Ĺ—č –ļ –ď–Ľ–ł–≤–Ķ–ļ—É. –Ě–Ķ—Ā–ľ–ĺ—ā—Ä—Ź –Ĺ–į –≤—č—Ā–ĺ–ļ—É—é —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć —ā–Ķ—Ä–į–Ņ–ł–ł –ď–Ľ–ł–≤–Ķ–ļ–ĺ–ľ, –ľ–ł–Ĺ–ĺ—Ä–Ĺ–į—Ź —á–į—Ā—ā—Ć –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö –ī–Ķ–ľ–ĺ–Ĺ—Ā—ā—Ä–ł—Ä—É–Ķ—ā –Ņ–Ķ—Ä–≤–ł—á–Ĺ—É—é —Ä–Ķ–∑–ł—Ā—ā–Ķ–Ĺ—ā–Ĺ–ĺ—Ā—ā—Ć, –į —É –Ī–ĺ–Ľ—Ć—ą–ł–Ĺ—Ā—ā–≤–į –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ –ī–į–∂–Ķ –Ņ–ĺ—Ā–Ľ–Ķ –ľ–Ĺ–ĺ–≥–ĺ–Ľ–Ķ—ā–Ĺ–Ķ–Ļ —É—Ā–Ņ–Ķ—ą–Ĺ–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł –Ĺ–į—Ā—ā—É–Ņ–į–Ķ—ā –≤—ā–ĺ—Ä–ł—á–Ĺ–į—Ź —Ä–Ķ–∑–ł—Ā—ā–Ķ–Ĺ—ā–Ĺ–ĺ—Ā—ā—Ć. –í –ļ–į—á–Ķ—Ā—ā–≤–Ķ –≤—č–Ī–ĺ—Ä–į –Ņ—Ä–ł –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł–ł –Ņ–į—ā–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –Ņ—Ä–ĺ—Ü–Ķ—Ā—Ā–į –ī–ĺ–∑—É –ď–Ľ–ł–≤–Ķ–ļ–į –Ņ–ĺ–≤—č—ą–į—é—ā —Ā 400 –ī–ĺ 800 –ľ–≥ –Ľ–ł–Ī–ĺ –ĺ—Ā—É—Č–Ķ—Ā—ā–≤–Ľ—Ź—é—ā –Ņ–Ķ—Ä–Ķ—Ö–ĺ–ī –Ĺ–į –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā –≤—ā–ĺ—Ä–ĺ–Ļ –Ľ–ł–Ĺ–ł–ł –°—É—ā–Ķ–Ĺ—ā. –≠—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć –°—É—ā–Ķ–Ĺ—ā–į –Ņ–ĺ—Ā–Ľ–Ķ –Ņ—Ä–ł–Ķ–ľ–į –ď–Ľ–ł–≤–Ķ–ļ–į —Ā–ĺ—Ā—ā–į–≤–Ľ—Ź–Ķ—ā –ĺ–ļ–ĺ–Ľ–ĺ 3‚Äď4 –ľ–Ķ—Ā. –ł, –ļ–į–ļ –ĺ–ļ–į–∑–į–Ľ–ĺ—Ā—Ć, —ā–į–ļ–∂–Ķ –∑–į–≤–ł—Ā–ł—ā –ĺ—ā –ľ—É—ā–į—Ü–ł–ĺ–Ĺ–Ĺ–ĺ–≥–ĺ —Ā—ā–į—ā—É—Ā–į. –í –ļ–į—á–Ķ—Ā—ā–≤–Ķ –ł–Ĺ–≥–ł–Ī–ł—ā–ĺ—Ä–ĺ–≤ —ā–ł—Ä–ĺ–∑–ł–Ĺ–ļ–ł–Ĺ–į–∑ —ā–į–ļ–∂–Ķ –ł—Ā—Ā–Ľ–Ķ–ī—É—é—ā—Ā—Ź –Ĺ–ł–Ľ–ĺ—ā–ł–Ĺ–ł–Ī, –ī–į–∑–į—ā–ł–Ĺ–ł–Ī, —Ā–ĺ—Ä–į—Ą–Ķ–Ĺ–ł–Ī, —Ä–Ķ–≥–ĺ—Ä–į—Ą–Ķ–Ĺ–ł–Ī, –≤–į—ā–į–Ľ–į–Ĺ–ł–Ī, –ľ–į–∑–ł—ā–ł–Ĺ–ł–Ī, –Ņ–į–∑–ĺ–Ņ–į–Ĺ–ł–Ī, –ļ—Ä–Ķ–Ĺ–ĺ–Ľ–į–Ĺ–ł–Ī. –†—Ź–ī –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤ —Ā –ł–Ĺ—č–ľ –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ–ĺ–ľ –ī–Ķ–Ļ—Ā—ā–≤–ł—Ź –Ņ—Ä–ĺ—Ö–ĺ–ī—Ź—ā –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź II —Ą–į–∑—č [1, 2].
–ě—Ā–Ĺ–ĺ–≤–Ĺ–ĺ–Ļ –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ –Ņ–į—ā–ĺ–≥–Ķ–Ĺ–Ķ–∑–į —Ä–į–∑–≤–ł—ā–ł—Ź GIST ‚Äď –ľ—É—ā–į—Ü–ł–ł –≤ –≥–Ķ–Ĺ–į—Ö c-kit –ł —ā—Ä–ĺ–ľ–Ī–ĺ—Ü–ł—ā–į—Ä–Ĺ–ĺ–≥–ĺ —Ą–į–ļ—ā–ĺ—Ä–į —Ä–ĺ—Ā—ā–į (PDGFRA)
–í 1998 –≥. S. Hirota –ł —Ā–ĺ–į–≤—ā. –ĺ–Ņ—É–Ī–Ľ–ł–ļ–ĺ–≤–į–Ľ–ł –≤ –∂—É—Ä–Ĺ–į–Ľ–Ķ Science —Ā—ā–į—ā—Ć—é ¬ę–Ē–ĺ–ľ–ł–Ĺ–į–Ĺ—ā–Ĺ–ĺ-–Ĺ–Ķ–≥–į—ā–ł–≤–Ĺ—č–Ķ –ľ—É—ā–į—Ü–ł–ł c-kit –Ņ—Ä–ł –≥–į—Ā—ā—Ä–ĺ–ł–Ĺ—ā–Ķ—Ā—ā–ł–Ĺ–į–Ľ—Ć–Ĺ—č—Ö —Ā—ā—Ä–ĺ–ľ–į–Ľ—Ć–Ĺ—č—Ö –ĺ–Ņ—É—Ö–ĺ–Ľ—Ź—Ö —É —á–Ķ–Ľ–ĺ–≤–Ķ–ļ–į¬Ľ [3]. –°—Ä–Ķ–ī–ł 58¬†–ĺ–Ņ—É—Ö–ĺ–Ľ–Ķ–Ļ –ľ–Ķ–∑–Ķ–Ĺ—Ö–ł–ľ–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–∂–ī–Ķ–Ĺ–ł—Ź —Ā –Ņ–ĺ–ľ–ĺ—Č—Ć—é –ł–ľ–ľ—É–Ĺ–ĺ–≥–ł—Ā—ā–ĺ—Ö–ł–ľ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –ľ–Ķ—ā–ĺ–ī–į –Ī—č–Ľ–ĺ –≤—č—Ź–≤–Ľ–Ķ–Ĺ–ĺ 49 GIST. –Ę–į–ļ–∂–Ķ –Ī—č–Ľ–ĺ –Ņ–ĺ–ļ–į–∑–į–Ĺ–ĺ, —á—ā–ĺ —ć–ļ—Ā–Ņ—Ä–Ķ—Ā—Ā–ł—Ź CD117 –Ĺ–į–Ī–Ľ—é–ī–į–Ķ—ā—Ā—Ź –Ĺ–Ķ —ā–ĺ–Ľ—Ć–ļ–ĺ –Ĺ–į –ļ–Ľ–Ķ—ā–ļ–į—Ö –ĺ–Ņ—É—Ö–ĺ–Ľ–ł, –Ĺ–ĺ –ł –Ĺ–į –ļ–Ľ–Ķ—ā–ļ–į—Ö –ö–į—Ö–į–Ľ–į, —á—ā–ĺ –Ņ–ĺ–∑–≤–ĺ–Ľ–ł–Ľ–ĺ –≤—č—Ā–ļ–į–∑–į—ā—Ć –Ņ—Ä–Ķ–ī–Ņ–ĺ–Ľ–ĺ–∂–Ķ–Ĺ–ł–Ķ –ĺ –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–∂–ī–Ķ–Ĺ–ł–ł GIST –ł–ľ–Ķ–Ĺ–Ĺ–ĺ –ł–∑ —ć—ā–ł—Ö –ļ–Ľ–Ķ—ā–ĺ–ļ. –í 5 –ł–∑ 6 –ĺ–Ī—Ä–į–∑—Ü–ĺ–≤ –ĺ–Ņ—É—Ö–ĺ–Ľ–Ķ–Ļ –į–≤—ā–ĺ—Ä—č –≤—č—Ź–≤–ł–Ľ–ł –ľ—É—ā–į—Ü–ł—é –≤ –≥–Ķ–Ĺ–Ķ c-kit. –í –Ņ–ĺ—Ā–Ľ–Ķ–ī—É—é—Č–Ķ–ľ –Ņ–ĺ–ī—ā–≤–Ķ—Ä–ī–ł–Ľ–ĺ—Ā—Ć, —á—ā–ĺ –ľ—É—ā–į—Ü–ł—Ź –≥–Ķ–Ĺ–į c-kit ‚Äď —ć—ā–ĺ –ĺ—Ā–Ĺ–ĺ–≤–Ĺ–ĺ–Ļ –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ —Ä–į–∑–≤–ł—ā–ł—Ź GIST. –í —Ā–Ķ–Ĺ—ā—Ź–Ī—Ä–Ķ 1998 –≥. –≤ –•–Ķ–Ľ—Ć—Ā–ł–Ĺ–ļ–ł, –§–ł–Ĺ–Ľ—Ź–Ĺ–ī–ł—Ź, –Ņ—Ä–ĺ—ą–Ķ–Ľ –Ņ–Ķ—Ä–≤—č–Ļ –ľ–Ķ–∂–ī—É–Ĺ–į—Ä–ĺ–ī–Ĺ—č–Ļ —Ā–ł–ľ–Ņ–ĺ–∑–ł—É–ľ –Ņ–ĺ –ł–∑—É—á–Ķ–Ĺ–ł—é GIST, –Ĺ–į –ļ–ĺ—ā–ĺ—Ä–ĺ–ľ –Ņ—Ä–ł—Ā—É—ā—Ā—ā–≤–ĺ–≤–į–Ľ–ł 23 –ī–Ķ–Ľ–Ķ–≥–į—ā–į. –Ě–į —Ā–ł–ľ–Ņ–ĺ–∑–ł—É–ľ–Ķ –Ī—č–Ľ–ĺ –≤—č—Ā–ļ–į–∑–į–Ĺ–ĺ –Ņ—Ä–Ķ–ī–Ņ–ĺ–Ľ–ĺ–∂–Ķ–Ĺ–ł–Ķ –ĺ –Ņ–ĺ—ā–Ķ–Ĺ—Ü–ł–į–Ľ—Ć–Ĺ–ĺ–Ļ —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł –ł–Ĺ–≥–ł–Ī–ł—ā–ĺ—Ä–ĺ–≤ c-kit –Ņ—Ä–ł GIST.
–Ď–Ķ–Ľ–ĺ–ļ KIT, –ļ–ĺ–ī–ł—Ä—É–Ķ–ľ—č–Ļ –≥–Ķ–Ĺ–ĺ–ľ c-kit, —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź —á–Ľ–Ķ–Ĺ–ĺ–ľ —Ā–Ķ–ľ–Ķ–Ļ—Ā—ā–≤–į —ā–ł—Ä–ĺ–∑–ł–Ĺ–ļ–ł–Ĺ–į–∑. –í —ć—ā—É –∂–Ķ –≥—Ä—É–Ņ–Ņ—É –≤—Ö–ĺ–ī—Ź—ā –į–Ľ—Ć—Ą–į- –ł –Ī–Ķ—ā–į-—Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä—č —ā—Ä–ĺ–ľ–Ī–ĺ—Ü–ł—ā–į—Ä–Ĺ–ĺ–≥–ĺ —Ą–į–ļ—ā–ĺ—Ä–į —Ä–ĺ—Ā—ā–į (PDGFRA –ł PDRGFRB), —Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä –ļ–ĺ–Ľ–ĺ–Ĺ–ł–Ķ—Ā—ā–ł–ľ—É–Ľ–ł—Ä—É—é—Č–Ķ–≥–ĺ —Ą–į–ļ—ā–ĺ—Ä–į –ľ–į–ļ—Ä–ĺ—Ą–į–≥–ĺ–≤ (CSF1R) –ł —Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä —Ü–ł—ā–ĺ–ļ–ł–Ĺ–į Fl. –õ–ł–≥–į–Ĺ–ī–ĺ–ľ –ī–Ľ—Ź KIT —Ā–Ľ—É–∂–ł—ā —Ā—ā–≤–ĺ–Ľ–ĺ–≤—č–Ļ –ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—č–Ļ —Ą–į–ļ—ā–ĺ—Ä (SCF). –†–Ķ–∑—É–Ľ—Ć—ā–į—ā–į–ľ–ł –≤–∑–į–ł–ľ–ĺ–ī–Ķ–Ļ—Ā—ā–≤–ł—Ź —Ź–≤–Ľ—Ź—é—ā—Ā—Ź –≥–ĺ–ľ–ĺ–ī–ł–ľ–Ķ—Ä–ł–∑–į—Ü–ł—Ź —Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–į –ł –∑–į–Ņ—É—Ā–ļ –ļ–ł–Ĺ–į–∑–Ĺ–ĺ–Ļ –į–ļ—ā–ł–≤–į—Ü–ł–ł. –ü—Ä–ł —Ä–į–∑–≤–ł—ā–ł–ł GIST –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–ī–ł—ā –Ĺ–Ķ–∑–į–≤–ł—Ā–ł–ľ–į—Ź –ĺ—ā –Ĺ–į–Ľ–ł—á–ł—Ź –Ľ–ł–≥–į–Ĺ–ī–į –į–ļ—ā–ł–≤–į—Ü–ł—Ź, —á—ā–ĺ –ł –Ņ—Ä–ł–≤–ĺ–ī–ł—ā –ļ —Ä–į–∑–≤–ł—ā–ł—é –ĺ–Ņ—É—Ö–ĺ–Ľ–ł. –Ě–į–ł–Ī–ĺ–Ľ–Ķ–Ķ —á–į—Ā—ā–į—Ź –ľ—É—ā–į—Ü–ł—Ź –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–ī–ł—ā –≤ 11-–ľ —ć–ļ–∑–ĺ–Ĺ–Ķ –≥–Ķ–Ĺ–į, –ļ–ĺ—ā–ĺ—Ä—č–Ļ –ļ–ĺ–ī–ł—Ä—É–Ķ—ā –≤–Ĺ—É—ā—Ä–ł–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—č–Ļ –ī–ĺ–ľ–Ķ–Ĺ. –ü—Ä–ł —ć—ā–ĺ–ľ –≤–ĺ–∑–ľ–ĺ–∂–Ĺ—č —Ä–į–∑–Ľ–ł—á–Ĺ—č–Ķ —ā–ł–Ņ—č –ľ—É—ā–į—Ü–ł–Ļ: –ī–Ķ–Ľ–Ķ—Ü–ł–ł, –≤—Ā—ā–į–≤–ļ–ł, –∑–į–ľ–Ķ–Ĺ—č –ł–Ľ–ł –ļ–ĺ–ľ–Ī–ł–Ĺ–į—Ü–ł–ł. –° –ī–Ķ–Ľ–Ķ—Ü–ł—Ź–ľ–ł —Ā–≤—Ź–∑–į–Ĺ —Ö—É–ī—ą–ł–Ļ –Ņ—Ä–ĺ–≥–Ĺ–ĺ–∑, –ī–Ľ—Ź –ī—Ä—É–≥–ł—Ö —ā–ł–Ņ–ĺ–≤ –ľ—É—ā–į—Ü–ł–Ļ —Ö–į—Ä–į–ļ—ā–Ķ—Ä–Ĺ–ĺ –Ī–ĺ–Ľ–Ķ–Ķ –Ī–Ľ–į–≥–ĺ–Ņ—Ä–ł—Ź—ā–Ĺ–ĺ–Ķ —ā–Ķ—á–Ķ–Ĺ–ł–Ķ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź [4, 5].
–ü—Ä–ł–Ī–Ľ–ł–∑–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ –≤ 10% —Ā–Ľ—É—á–į–Ķ–≤ –ľ—É—ā–į—Ü–ł–ł –Ņ—Ä–ĺ–ł—Ā—Ö–ĺ–ī—Ź—ā –≤ 9-–ľ —ć–ļ–∑–ĺ–Ĺ–Ķ –≥–Ķ–Ĺ–į c-kit, –ļ–ĺ—ā–ĺ—Ä—č–Ļ –ļ–ĺ–ī–ł—Ä—É–Ķ—ā –≤–Ĺ–Ķ–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—č–Ļ –ī–ĺ–ľ–Ķ–Ĺ –Ī–Ķ–Ľ–ļ–į. –ö–ł–Ĺ–į–∑–Ĺ—č–Ļ –ī–ĺ–ľ–Ķ–Ĺ –Ņ—Ä–ł –ľ—É—ā–į—Ü–ł–ł –≤ 9-–ľ —ć–ļ–∑–ĺ–Ĺ–Ķ –Ņ–ĺ—Ö–ĺ–∂ –Ĺ–į –ļ–ł–Ĺ–į–∑–Ĺ—č–Ļ –ī–ĺ–ľ–Ķ–Ĺ –Ņ—Ä–ł –ī–ł–ļ–ĺ–ľ —ā–ł–Ņ–Ķ GIST, —á—ā–ĺ –Ņ—Ä–Ķ–ī–Ņ–ĺ–Ľ–į–≥–į–Ķ—ā —á—É–≤—Ā—ā–≤–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ—Ā—ā—Ć –ļ —ā–Ķ—Ä–į–Ņ–ł–ł –Ņ–ĺ—Ā–Ľ–Ķ–ī–Ĺ–ł—Ö. –ė–Ĺ—ā–Ķ—Ä–Ķ—Ā–Ĺ–ĺ, —á—ā–ĺ –ľ—É—ā–į—Ü–ł–ł –≤ 9-–ľ —ć–ļ–∑–ĺ–Ĺ–Ķ –≤—Ā—ā—Ä–Ķ—á–į—é—ā—Ā—Ź –Ņ—Ä–į–ļ—ā–ł—á–Ķ—Ā–ļ–ł –ł—Ā–ļ–Ľ—é—á–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ –≤ –ĺ–Ņ—É—Ö–ĺ–Ľ—Ź—Ö —ā–ĺ–Ĺ–ļ–ĺ–Ļ –ł —ā–ĺ–Ľ—Ā—ā–ĺ–Ļ –ļ–ł—ą–ļ–ł, –≤ –ĺ–Ņ—É—Ö–ĺ–Ľ—Ź—Ö –∂–Ķ–Ľ—É–ī–ļ–į —á–į—Ā—ā–ĺ—ā–į –ľ—É—ā–į—Ü–ł–ł ‚Äď –ĺ–ļ–ĺ–Ľ–ĺ 1% [6]. –ú—É—ā–į—Ü–ł–ł –≤ —ć–ļ–∑–ĺ–Ĺ–į—Ö 13 –ł 17 —Ā–ĺ—Ā—ā–į–≤–Ľ—Ź—é—ā 1‚Äď2% —Ā–Ľ—É—á–į–Ķ–≤. –ü—Ä–Ķ–ī–Ņ–ĺ–Ľ–į–≥–į–Ķ—ā—Ā—Ź, —á—ā–ĺ –ī–į–Ĺ–Ĺ—č–Ķ —ć–ļ–∑–ĺ–Ĺ—č –ĺ—ā–≤–Ķ—á–į—é—ā –∑–į —Ą–ł–∑–ł–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ĺ–Ķ –Ņ–ĺ–ī–į–≤–Ľ–Ķ–Ĺ–ł–Ķ —Ą—É–Ĺ–ļ—Ü–ł–ł –≤–Ĺ—É—ā—Ä–ł–ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—č—Ö –ī–ĺ–ľ–Ķ–Ĺ–ĺ–≤. –ě—á–Ķ–≤–ł–ī–Ĺ–ĺ, —á—ā–ĺ –ľ—É—ā–į—Ü–ł—Ź –≥–Ķ–Ĺ–į —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –ļ–Ľ—é—á–Ķ–≤–ĺ–Ļ –ł –Ĺ–į—á–į–Ľ—Ć–Ĺ–ĺ–Ļ —Ā—ā–į–ī–ł–Ķ–Ļ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź. –í –ī–į–Ľ—Ć–Ĺ–Ķ–Ļ—ą–Ķ–ľ –∑–į–Ņ—É—Ā–ļ–į–Ķ—ā—Ā—Ź –ļ–į—Ā–ļ–į–ī —Ā–į–ľ—č—Ö —Ä–į–∑–Ľ–ł—á–Ĺ—č—Ö —Ā–ł–≥–Ĺ–į–Ľ—Ć–Ĺ—č—Ö –Ņ—É—ā–Ķ–Ļ (MAPK, PI3K-AKT, MYC, mTOR). –ė—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź –Ĺ–į –ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—č—Ö –Ľ–ł–Ĺ–ł—Ź—Ö –Ņ–ĺ–ļ–į–∑–į–Ľ–ł, —á—ā–ĺ, –≤ —á–į—Ā—ā–Ĺ–ĺ—Ā—ā–ł, –Ņ–ĺ–ī–į–≤–Ľ–Ķ–Ĺ–ł–Ķ –Ņ—É—ā–Ķ–Ļ PI3K –Ī–ĺ–Ľ–Ķ–Ķ —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ –Ņ–ĺ —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—é —Ā –ł–Ĺ–≥–ł–Ī–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ–ľ –ú–ź–†–ö –ł mTOR [7].
–ú—É—ā–į—Ü–ł–ł –≤ –≥–Ķ–Ĺ–Ķ —ā—Ä–ĺ–ľ–Ī–ĺ—Ü–ł—ā–į—Ä–Ĺ–ĺ–≥–ĺ —Ą–į–ļ—ā–ĺ—Ä–į —Ä–ĺ—Ā—ā–į –≤—Ā—ā—Ä–Ķ—á–į—é—ā—Ā—Ź –≤ 5‚Äď10% —Ā–Ľ—É—á–į–Ķ–≤. –ö–į–ļ –ł –≤ —Ā–Ľ—É—á–į–Ķ —Ā –į–ļ—ā–ł–≤–į—Ü–ł–Ķ–Ļ —Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–į KIT –Ĺ–į –ľ–Ķ–ľ–Ī—Ä–į–Ĺ–Ķ –ļ–Ľ–Ķ—ā–ĺ–ļ, —Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä PDGFRA –Ĺ–į—á–ł–Ĺ–į–Ķ—ā –į–ļ—ā–ł–≤–ł—Ä–ĺ–≤–į—ā—Ć—Ā—Ź –Ī–Ķ–∑ —Ā–≤–ĺ–Ķ–≥–ĺ –Ľ–ł–≥–į–Ĺ–ī–į PDGFA, –≤ —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā–Ķ —á–Ķ–≥–ĺ –∑–į–Ņ—É—Ā–ļ–į–Ķ—ā—Ā—Ź –ļ–į—Ā–ļ–į–ī —Ä–Ķ–į–ļ—Ü–ł–Ļ, —Ā—Ö–ĺ–∂–ł—Ö —Ā —Ä–Ķ–į–ļ—Ü–ł—Ź–ľ–ł, –ł–ľ–Ķ—é—Č–ł–ľ–ł –ľ–Ķ—Ā—ā–ĺ –Ņ—Ä–ł –į–ļ—ā–ł–≤–į—Ü–ł–ł KIT [8]. –Ē–Ľ—Ź –Ņ–ĺ–ī—ā–≤–Ķ—Ä–∂–ī–Ķ–Ĺ–ł—Ź –ī–ł–į–≥–Ĺ–ĺ–∑–į GIST –Ņ–į–Ĺ–Ķ–Ľ—Ć –ł–ľ–ľ—É–Ĺ–ĺ–≥–ł—Ā—ā–ĺ—Ö–ł–ľ–ł—á–Ķ—Ā–ļ–ĺ–Ļ —Ä–Ķ–į–ļ—Ü–ł–ł –≤–ļ–Ľ—é—á–į–Ķ—ā, –ļ—Ä–ĺ–ľ–Ķ CD117, –ľ–į—Ä–ļ–Ķ—Ä—č DOG1 –ł PKC-—ā–Ķ—ā–į [9, 10].
–í 10‚Äď15% —Ā–Ľ—É—á–į–Ķ–≤ GIST –Ĺ–Ķ —É–ī–į–Ķ—ā—Ā—Ź –≤—č—Ź–≤–ł—ā—Ć –ľ—É—ā–į—Ü–ł–ł –≤ –≥–Ķ–Ĺ–į—Ö c-kit –ł PDGFRA, —ā–į–ļ–ł–ľ –ĺ–Ī—Ä–į–∑–ĺ–ľ, —Ä–Ķ—á—Ć –ł–ī–Ķ—ā –ĺ –ī–ł–ļ–ĺ–ľ —ā–ł–Ņ–Ķ —ć—ā–ł—Ö –≥–Ķ–Ĺ–ĺ–≤. –í —ā–ĺ –∂–Ķ –≤—Ä–Ķ–ľ—Ź –ł–ľ–ľ—É–Ĺ–ĺ–≥–ł—Ā—ā–ĺ—Ö–ł–ľ–ł—á–Ķ—Ā–ļ–ł –ĺ–Ņ—Ä–Ķ–ī–Ķ–Ľ—Ź–Ķ—ā—Ā—Ź —ć–ļ—Ā–Ņ—Ä–Ķ—Ā—Ā–ł—Ź CD117, —á—ā–ĺ –≥–ĺ–≤–ĺ—Ä–ł—ā –ĺ –∑–į–Ņ—É—Ā–ļ–Ķ —Ā–ł–≥–Ĺ–į–Ľ—Ć–Ĺ—č—Ö –Ņ—É—ā–Ķ–Ļ, –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ –ļ–ĺ—ā–ĺ—Ä–ĺ–≥–ĺ –Ĺ–Ķ —Ā–ĺ–≤—Ā–Ķ–ľ —Ź—Ā–Ķ–Ĺ. –í –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ļ –Ņ—Ä–į–ļ—ā–ł–ļ–Ķ —á–į—Ā—ā–ĺ –Ņ—Ä–ĺ–≤–ĺ–ī–ł—ā—Ā—Ź —Ā–ļ—Ä–ł–Ĺ–ł–Ĺ–≥ —ā–ĺ–Ľ—Ć–ļ–ĺ –Ĺ–į –ľ—É—ā–į—Ü–ł–ł –≤ 11-–ľ –ł 9-–ľ —ć–ļ–∑–ĺ–Ĺ–į—Ö, –Ņ—Ä–ł –ĺ—ā—Ā—É—ā—Ā—ā–≤–ł–ł –ľ—É—ā–į—Ü–ł–ł —Ą–ĺ—Ä–ľ–į–Ľ—Ć–Ĺ–ĺ –Ņ—Ä–Ķ–ī–Ņ–ĺ–Ľ–į–≥–į–Ķ—ā—Ā—Ź –ī–ł–ļ–ł–Ļ —ā–ł–Ņ, –į –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ķ –Ĺ–į –ľ—É—ā–į—Ü–ł—é –≤ 13-–ľ –ł–Ľ–ł 17-–ľ —ć–ļ–∑–ĺ–Ĺ–į—Ö –Ĺ–Ķ –Ņ—Ä–ĺ–≤–ĺ–ī–ł—ā—Ā—Ź. –ú–ĺ—Ä—Ą–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ł –ł –ľ–į–ļ—Ä–ĺ—Ā–ļ–ĺ–Ņ–ł—á–Ķ—Ā–ļ–ł –ĺ–Ņ—É—Ö–ĺ–Ľ–ł —Ā –ī–ł–ļ–ł–ľ —ā–ł–Ņ–ĺ–ľ –Ĺ–ł—á–Ķ–ľ –Ĺ–Ķ –ĺ—ā–Ľ–ł—á–į—é—ā—Ā—Ź –ĺ—ā –ľ—É—ā–į–Ĺ—ā–Ĺ—č—Ö –ĺ–Ņ—É—Ö–ĺ–Ľ–Ķ–Ļ. –í —ā–ĺ –∂–Ķ –≤—Ä–Ķ–ľ—Ź —ā—Č–į—ā–Ķ–Ľ—Ć–Ĺ—č–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź –Ņ–ĺ–ļ–į–∑–į–Ľ–ł, —á—ā–ĺ –Ņ—Ä–ł –ī–ł–ļ–ĺ–ľ —ā–ł–Ņ–Ķ –≤—č—Ź–≤–Ľ—Ź—é—ā—Ā—Ź –ī—Ä—É–≥–ł–Ķ –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ—č –į–ļ—ā–ł–≤–į—Ü–ł–ł –ł –Ņ—Ä–ĺ–≥–Ĺ–ĺ–∑ —ā–Ķ—á–Ķ–Ĺ–ł—Ź –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź –Ņ—Ä–ł —ć—ā–ł—Ö –ĺ–Ņ—É—Ö–ĺ–Ľ—Ź—Ö —ā–į–ļ–∂–Ķ –ĺ—ā–Ľ–ł—á–Ķ–Ĺ –ĺ—ā GIST —Ā –ľ—É—ā–į—Ü–ł—Ź–ľ–ł. –Ě–į–ł–Ī–ĺ–Ľ–Ķ–Ķ —á–į—Ā—ā–ĺ (–≤ –Ņ–ĺ–Ľ–ĺ–≤–ł–Ĺ–Ķ —Ā–Ľ—É—á–į–Ķ–≤) –Ņ—Ä–ł –ī–ł–ļ–ĺ–ľ —ā–ł–Ņ–Ķ –≤—č—Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –Ņ–ĺ–≤—č—ą–Ķ–Ĺ–Ĺ–į—Ź —ć–ļ—Ā–Ņ—Ä–Ķ—Ā—Ā–ł—Ź —Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–į –ł–Ĺ—Ā—É–Ľ–ł–Ĺ–ĺ–≤–ĺ–≥–ĺ —Ą–į–ļ—ā–ĺ—Ä–į —Ä–ĺ—Ā—ā–į (IGF1R), –ļ–ĺ—ā–ĺ—Ä—č–Ļ –ī–Ķ–Ļ—Ā—ā–≤—É–Ķ—ā —á–Ķ—Ä–Ķ–∑ —Ā–ł–≥–Ĺ–į–Ľ—Ć–Ĺ—č–Ķ –Ņ—É—ā–ł –ú–ź–†–ö –ł PI3-AKT.¬†
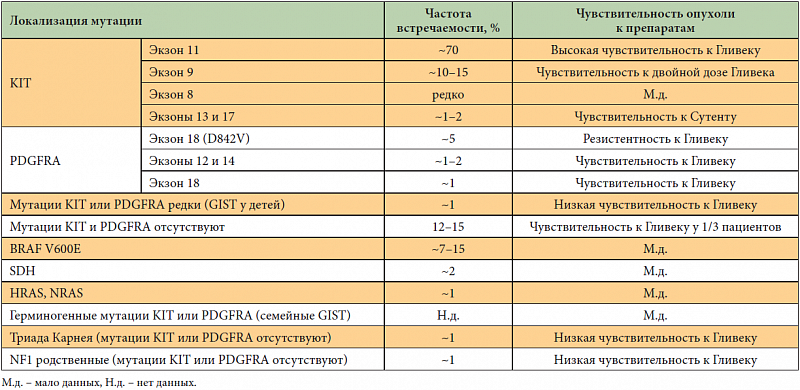
–ė–Ĺ—ā–Ķ—Ä–Ķ—Ā–Ĺ–ĺ, —á—ā–ĺ –Ņ–ĺ–≤—č—ą–Ķ–Ĺ–Ĺ–į—Ź —ć–ļ—Ā–Ņ—Ä–Ķ—Ā—Ā–ł—Ź IGF1R –≤—č—Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –Ņ—Ä–ł –∑–į–Ī—Ä—é—ą–ł–Ĺ–Ĺ—č—Ö —Ā–į—Ä–ļ–ĺ–ľ–į—Ö –ł —Ā–į—Ä–ļ–ĺ–ľ–Ķ –ģ–ł–Ĺ–≥–į. –ú—É—ā–į—Ü–ł–ł –≤ –≥–Ķ–Ĺ–Ķ BRAF, –ļ–ĺ—ā–ĺ—Ä—č–Ķ –Ī–ĺ–Ľ–Ķ–Ķ —Ö–į—Ä–į–ļ—ā–Ķ—Ä–Ĺ—č –ī–Ľ—Ź –ľ–Ķ–Ľ–į–Ĺ–ĺ–ľ—č –ł —Ä–į–ļ–į —Č–ł—ā–ĺ–≤–ł–ī–Ĺ–ĺ–Ļ –∂–Ķ–Ľ–Ķ–∑—č, –≤—č—Ź–≤–Ľ—Ź—é—ā—Ā—Ź –Ņ—Ä–ł –ī–ł–ļ–ĺ–ľ —ā–ł–Ņ–Ķ GIST –≤ 13% —Ā–Ľ—É—á–į–Ķ–≤ [11]. –í —Ä—Ź–ī–Ķ —Ā–Ľ—É—á–į–Ķ–≤ –Ņ—Ä–ł –ī–ł–ļ–ĺ–ľ —ā–ł–Ņ–Ķ GIST —É–ī–į–Ķ—ā—Ā—Ź –ĺ–Ī–Ĺ–į—Ä—É–∂–ł—ā—Ć –ľ—É—ā–į—Ü–ł—é –≤ –≥–Ķ–Ĺ–Ķ —Ā—É–ļ—Ü–ł–Ĺ–į—ā–ī–Ķ–≥–ł–ī—Ä–ĺ–≥–Ķ–Ĺ–į–∑—č (SDH), –ļ–ĺ—ā–ĺ—Ä—č–Ļ –ļ–ĺ–ī–ł—Ä—É–Ķ—ā —á–Ķ—ā—č—Ä–Ķ —Ā—É–Ī—ä–Ķ–ī–ł–Ĺ–ł—Ü—č (SDHA, SDHB, SDHC, SDHD). –§—É–Ĺ–ļ—Ü–ł—Ź —ć—ā–ĺ–≥–ĺ —Ą–Ķ—Ä–ľ–Ķ–Ĺ—ā–į ‚Äď —É—á–į—Ā—ā–ł–Ķ –≤ —Ü–ł–ļ–Ľ–Ķ –ö—Ä–Ķ–Ī—Ā–į. –ú—É—ā–į—Ü–ł—Ź —É–ļ–į–∑–į–Ĺ–Ĺ–ĺ–≥–ĺ –≥–Ķ–Ĺ–į –Ņ–ĺ–≤—č—ą–į–Ķ—ā —Ä–ł—Ā–ļ —Ä–į–∑–≤–ł—ā–ł—Ź –Ĺ–Ķ —ā–ĺ–Ľ—Ć–ļ–ĺ GIST, –Ĺ–ĺ –ł —Ā–ł–Ĺ—Ö—Ä–ĺ–Ĺ–Ĺ—č—Ö –Ņ–į—Ä–į–≥–į–Ĺ–≥–Ľ–ł–ĺ–ľ (—Ā–ł–Ĺ–ī—Ä–ĺ–ľ –ö–į—Ä–Ĺ–Ķ—Ź ‚Äď –°—ā—Ä–į—ā–į–ļ–ł—Ā–į, Carney ‚Äď Stratakis syndrome) [12]. –ú–Ķ—Ö–į–Ĺ–ł–∑–ľ —Ä–į–∑–≤–ł—ā–ł—Ź GIST –Ņ—Ä–ł –ľ—É—ā–į—Ü–ł–ł –≥–Ķ–Ĺ–į —Ā—É–ļ—Ü–ł–Ĺ–į—ā–ī–Ķ–≥–ł–ī—Ä–ĺ–≥–Ķ–Ĺ–į–∑—č –ĺ—Ā—ā–į–Ķ—ā—Ā—Ź –Ĺ–Ķ —Ā–ĺ–≤—Ā–Ķ–ľ —Ź—Ā–Ĺ—č–ľ, –Ĺ–ĺ –Ņ—Ä–Ķ–ī–Ņ–ĺ–Ľ–į–≥–į–Ķ—ā—Ā—Ź, —á—ā–ĺ —Ą–Ķ—Ä–ľ–Ķ–Ĺ—ā —Ā–Ĺ–ł–∂–į–Ķ—ā –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć –Ņ—Ä–ĺ–Ľ–ł–Ľ–≥–ł–ī—Ä–ĺ–ļ—Ā–ł–Ľ–į–∑—č, –ļ–ĺ—ā–ĺ—Ä–į—Ź, –≤ —Ā–≤–ĺ—é –ĺ—á–Ķ—Ä–Ķ–ī—Ć, —Ä–Ķ–≥—É–Ľ–ł—Ä—É–Ķ—ā –į–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć —Ą–į–ļ—ā–ĺ—Ä–į, –ł–Ĺ–ī—É—Ü–ł—Ä—É–Ķ–ľ–ĺ–≥–ĺ –≥–ł–Ņ–ĺ–ļ—Ā–ł–Ķ–Ļ (hypoxia-inducible factor, HIF1-–į–Ľ—Ć—Ą–į). –í —Ā–≤–ĺ—é –ĺ—á–Ķ—Ä–Ķ–ī—Ć HIF1-–į–Ľ—Ć—Ą–į —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź –į–ļ—ā–ł–≤–į—ā–ĺ—Ä–ĺ–ľ –ī–Ľ—Ź IGF2 –ł VEGF. –ö–Ľ–į—Ā—Ā–ł—Ą–ł–ļ–į—Ü–ł—Ź GIST –≤ –∑–į–≤–ł—Ā–ł–ľ–ĺ—Ā—ā–ł –ĺ—ā –Ľ–ĺ–ļ–į–Ľ–ł–∑–į—Ü–ł–ł –ľ—É—ā–į—Ü–ł–ł –ł —á—É–≤—Ā—ā–≤–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ—Ā—ā—Ć –ĺ–Ņ—É—Ö–ĺ–Ľ–Ķ–Ļ –ļ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į–ľ –Ņ—Ä–Ķ–ī—Ā—ā–į–≤–Ľ–Ķ–Ĺ—č –≤ —ā–į–Ī–Ľ–ł—Ü–Ķ 1.
–≠–Ņ–ł–ī–Ķ–ľ–ł–ĺ–Ľ–ĺ–≥–ł—Ź –≥–į—Ā—ā—Ä–ĺ–ł–Ĺ—ā–Ķ—Ā—ā–ł–Ĺ–į–Ľ—Ć–Ĺ—č—Ö —Ā—ā—Ä–ĺ–ľ–į–Ľ—Ć–Ĺ—č—Ö –ĺ–Ņ—É—Ö–ĺ–Ľ–Ķ–Ļ
–Ē–ĺ –ĺ—ā–ļ—Ä—č—ā–ł—Ź –ľ—É—ā–į—Ü–ł–ł –≥–Ķ–Ĺ–į c-kit –ł –ł—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į–Ĺ–ł—Ź –ľ–į—Ä–ļ–Ķ—Ä–į CD117 GIST —Ä–į—Ā—Ü–Ķ–Ĺ–ł–≤–į–Ľ–ł—Ā—Ć –ļ–į–ļ –Ľ–Ķ–Ļ–ĺ–ľ–ł–ĺ—Ā–į—Ä–ļ–ĺ–ľ—č. –°–ĺ–≥–Ľ–į—Ā–Ĺ–ĺ –ļ–Ľ–į—Ā—Ā–ł—Ą–ł–ļ–į—Ü–ł–ł –ł —Ā—ā–į–ī–ł—Ä–ĺ–≤–į–Ĺ–ł—é –∑–Ľ–ĺ–ļ–į—á–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ—č—Ö –ĺ–Ņ—É—Ö–ĺ–Ľ–Ķ–Ļ, –Ľ–Ķ–Ļ–ĺ–ľ–ł–ĺ—Ā–į—Ä–ļ–ĺ–ľ—č –∂–Ķ–Ľ—É–ī–ļ–į –ł —ā–ĺ–Ĺ–ļ–ĺ–Ļ –ļ–ł—ą–ļ–ł –Ī—č–Ľ–ł –ĺ—ā–Ĺ–Ķ—Ā–Ķ–Ĺ—č –ļ –∑–Ľ–ĺ–ļ–į—á–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ—č–ľ –ĺ–Ņ—É—Ö–ĺ–Ľ—Ź–ľ —É–ļ–į–∑–į–Ĺ–Ĺ—č—Ö –ĺ—Ä–≥–į–Ĺ–ĺ–≤ –ł, —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ, ¬ę—Ä–į—Ā—ā–≤–ĺ—Ä–ł–Ľ–ł—Ā—ƬĽ —Ā—Ä–Ķ–ī–ł –ī–ł–į–≥–Ĺ–ĺ–∑–ĺ–≤ ¬ę—Ä–į–ļ –∂–Ķ–Ľ—É–ī–ļ–į¬Ľ –ł –ĺ–Ņ—É—Ö–ĺ–Ľ–Ķ–Ļ —ā–ĺ–Ĺ–ļ–ĺ–Ļ –ļ–ł—ą–ļ–ł. –Ę–ĺ—á–Ĺ–į—Ź –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ķ–ľ–ĺ—Ā—ā—Ć –Ī—č–Ľ–į –Ĺ–Ķ–ł–∑–≤–Ķ—Ā—ā–Ĺ–į [13]. –ó–į –Ņ–ĺ—Ā–Ľ–Ķ–ī–Ĺ–ł–Ķ –≥–ĺ–ī—č –≤–ĺ –ľ–Ĺ–ĺ–≥–ł—Ö —Ā—ā—Ä–į–Ĺ–į—Ö, –ĺ—Ā–ĺ–Ī–Ķ–Ĺ–Ĺ–ĺ –≤ —ā–Ķ—Ö, –≥–ī–Ķ –≤–Ķ–ī—É—ā—Ā—Ź –ļ–į–Ĺ—Ü–Ķ—Ä-—Ä–Ķ–≥–ł—Ā—ā—Ä—č –ł —á–į—Ā—ā–ĺ —Ā–ĺ—Ö—Ä–į–Ĺ—Ź—é—ā—Ā—Ź –Ņ–į—Ä–į—Ą–ł–Ĺ–ĺ–≤—č–Ķ –Ī–Ľ–ĺ–ļ–ł –ł —Ā—ā–Ķ–ļ–Ľ–į —É–ī–į–Ľ–Ķ–Ĺ–Ĺ—č—Ö –ĺ–Ī—Ä–į–∑—Ü–ĺ–≤, —É–ī–į–Ľ–ĺ—Ā—Ć –Ņ—Ä–ĺ—Ź—Ā–Ĺ–ł—ā—Ć —ć–Ņ–ł–ī–Ķ–ľ–ł–ĺ–Ľ–ĺ–≥–ł—é GIST. –Ę–į–ļ, –Ĺ–į–ł–Ī–ĺ–Ľ–Ķ–Ķ –Ņ–ĺ–Ľ–Ĺ–ĺ—Ü–Ķ–Ĺ–Ĺ—č–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź –Ņ—Ä–ĺ–≤–Ķ–ī–Ķ–Ĺ—č –≤ —Ā–ļ–į–Ĺ–ī–ł–Ĺ–į–≤—Ā–ļ–ł—Ö —Ā—ā—Ä–į–Ĺ–į—Ö (–Ě–ĺ—Ä–≤–Ķ–≥–ł—Ź, –®–≤–Ķ—Ü–ł—Ź, –§–ł–Ĺ–Ľ—Ź–Ĺ–ī–ł—Ź, –Ē–į–Ĺ–ł—Ź) [14, 15]. –í —ć—ā–ł—Ö —Ā—ā—Ä–į–Ĺ–į—Ö —Ä–Ķ—ā—Ä–ĺ—Ā–Ņ–Ķ–ļ—ā–ł–≤–Ĺ–ĺ –Ī—č–Ľ–ł –Ņ–Ķ—Ä–Ķ—Ā–ľ–ĺ—ā—Ä–Ķ–Ĺ—č –Ī–Ľ–ĺ–ļ–ł ¬ę–Ľ–Ķ–Ļ–ĺ–ľ–ł–ĺ–ľ¬Ľ –ł ¬ę–Ľ–Ķ–Ļ–ĺ–ľ–ł–ĺ—Ā–į—Ä–ļ–ĺ–ľ¬Ľ –ł –≤ –Ņ–ĺ–ī–į–≤–Ľ—Ź—é—Č–Ķ–ľ –Ī–ĺ–Ľ—Ć—ą–ł–Ĺ—Ā—ā–≤–Ķ —Ā–Ľ—É—á–į–Ķ–≤ –ī–ł–į–≥–Ĺ–ĺ–∑ –Ī—č–Ľ –ł–∑–ľ–Ķ–Ĺ–Ķ–Ĺ –Ĺ–į GIST. –ě–ļ–į–∑–į–Ľ–ĺ—Ā—Ć, —á—ā–ĺ –≤ —Ā—Ä–Ķ–ī–Ĺ–Ķ–ľ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ķ–ľ–ĺ—Ā—ā—Ć —Ā–ĺ—Ā—ā–į–≤–Ľ—Ź–Ķ—ā 10‚Äď15 —á–Ķ–Ľ–ĺ–≤–Ķ–ļ –Ĺ–į 1 –ľ–Ľ–Ĺ –Ĺ–į—Ā–Ķ–Ľ–Ķ–Ĺ–ł—Ź. –í –ī—Ä—É–≥–ł—Ö –Ķ–≤—Ä–ĺ–Ņ–Ķ–Ļ—Ā–ļ–ł—Ö —Ā—ā—Ä–į–Ĺ–į—Ö —ć—ā–ĺ—ā –Ņ–ĺ–ļ–į–∑–į—ā–Ķ–Ľ—Ć –Ī—č–Ľ –Ĺ–ł–∂–Ķ, —á—ā–ĺ, —Ā–ļ–ĺ—Ä–Ķ–Ķ –≤—Ā–Ķ–≥–ĺ, –Ī—č–Ľ–ĺ —Ā–≤—Ź–∑–į–Ĺ–ĺ —Ā –ľ–Ķ–Ĺ—Ć—ą–ł–ľ –ĺ—Ö–≤–į—ā–ĺ–ľ –Ĺ–į—Ā–Ķ–Ľ–Ķ–Ĺ–ł—Ź —Ā–ļ—Ä–ł–Ĺ–ł–Ĺ–≥–ĺ–ľ –ł –Ī–ĺ–Ľ–Ķ–Ķ –Ĺ–ł–∑–ļ–ł–ľ —É—Ä–ĺ–≤–Ĺ–Ķ–ľ –ī–ĺ—Ā—ā—É–Ņ–Ĺ–ĺ—Ā—ā–ł –ľ–Ķ–ī–ł—Ü–ł–Ĺ—Ā–ļ–ĺ–Ļ –Ņ–ĺ–ľ–ĺ—Č–ł.¬†
–ö—Ä–ĺ–ľ–Ķ —ā–ĺ–≥–ĺ, —ā—Č–į—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ķ –ľ–ĺ—Ä—Ą–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ĺ–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ķ –ĺ–Ī—Ä–į–∑—Ü–ĺ–≤ –∂–Ķ–Ľ—É–ī–ļ–į —É –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö, —É–ľ–Ķ—Ä—ą–ł—Ö –ĺ—ā –ī—Ä—É–≥–ł—Ö –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł–Ļ, –Ņ–ĺ–∑–≤–ĺ–Ľ–ł–Ľ–ĺ –ĺ–Ī–Ĺ–į—Ä—É–∂–ł—ā—Ć —ā–į–ļ –Ĺ–į–∑—č–≤–į–Ķ–ľ—č–Ķ –ľ–ł–ļ—Ä–ĺ-GIST, —Ä–į–∑–ľ–Ķ—Ä–į–ľ–ł –ī–ĺ 1‚Äď2 –ľ–ľ. –í 2006 –≥. –≤ –∂—É—Ä–Ĺ–į–Ľ–Ķ Human Pathology –Ī—č–Ľ–į –ĺ–Ņ—É–Ī–Ľ–ł–ļ–ĺ–≤–į–Ĺ–į —Ā—ā–į—ā—Ć—Ź —Ź–Ņ–ĺ–Ĺ—Ā–ļ–ł—Ö –ľ–ĺ—Ä—Ą–ĺ–Ľ–ĺ–≥–ĺ–≤, –≤ –ļ–ĺ—ā–ĺ—Ä–ĺ–Ļ –≥–ĺ–≤–ĺ—Ä–ł–Ľ–ĺ—Ā—Ć –ĺ –≤—č—Ā–ĺ–ļ–ĺ–Ļ —á–į—Ā—ā–ĺ—ā–Ķ –≤—Ā—ā—Ä–Ķ—á–į–Ķ–ľ–ĺ—Ā—ā–ł GIST –≤ –∂–Ķ–Ľ—É–ī–ļ–Ķ [16]. –ź–≤—ā–ĺ—Ä—č –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ľ–ł 100 –∂–Ķ–Ľ—É–ī–ļ–ĺ–≤, —É–ī–į–Ľ–Ķ–Ĺ–Ĺ—č—Ö –Ņ–ĺ –Ņ–ĺ–≤–ĺ–ī—É —Ä–į–ļ–į. –í 35 –∂–Ķ–Ľ—É–ī–ļ–į—Ö –Ī—č–Ľ–ł –ĺ–Ī–Ĺ–į—Ä—É–∂–Ķ–Ĺ—č 50 –ľ–ł–ļ—Ä–ĺ—Ā–ļ–ĺ–Ņ–ł—á–Ķ—Ā–ļ–ł—Ö –ĺ–Ņ—É—Ö–ĺ–Ľ–Ķ–Ļ, –ļ–ĺ—ā–ĺ—Ä—č–Ķ –ĺ–ļ–į–∑–į–Ľ–ł—Ā—Ć –Ņ–ĺ–Ľ–ĺ–∂–ł—ā–Ķ–Ľ—Ć–Ĺ—č–ľ–ł –Ĺ–į —ć–ļ—Ā–Ņ—Ä–Ķ—Ā—Ā–ł—é KIT –ł/–ł–Ľ–ł CD34. –Ď–ĺ–Ľ—Ć—ą–ł–Ĺ—Ā—ā–≤–ĺ –ĺ–Ņ—É—Ö–ĺ–Ľ–Ķ–Ļ (90%) –Ľ–ĺ–ļ–į–Ľ–ł–∑–ĺ–≤–į–Ľ–ĺ—Ā—Ć –≤ –Ņ—Ä–ĺ–ļ—Ā–ł–ľ–į–Ľ—Ć–Ĺ—č—Ö –ĺ—ā–ī–Ķ–Ľ–į—Ö –∂–Ķ–Ľ—É–ī–ļ–į, –≤ 2¬†—Ā–Ľ—É—á–į—Ź—Ö GIST –≤—č—Ź–≤–Ľ–Ķ–Ĺ—č –ľ—É—ā–į—Ü–ł–ł –≥–Ķ–Ĺ–į c-kit. –í 28 –∂–Ķ–Ľ—É–ī–ļ–į—Ö –≤—č—Ź–≤–Ľ–Ķ–Ĺ–į 51 –Ľ–Ķ–Ļ–ĺ–ľ–ł–ĺ–ľ–į. –í 12 –∂–Ķ–Ľ—É–ī–ļ–į—Ö –ĺ–Ī–Ĺ–į—Ä—É–∂–Ķ–Ĺ–ĺ —Ā–ĺ—á–Ķ—ā–į–Ĺ–ł–Ķ –ľ–ł–ļ—Ä–ĺ—Ā–ļ–ĺ–Ņ–ł—á–Ķ—Ā–ļ–ł—Ö GIST –ł –Ľ–Ķ–Ļ–ĺ–ľ–ł–ĺ–ľ. –°—Ä–Ķ–ī–Ĺ–ł–Ļ —Ä–į–∑–ľ–Ķ—Ä –ĺ–Ņ—É—Ö–ĺ–Ľ–ł —Ā–ĺ—Ā—ā–į–≤–ł–Ľ 1,5 –ľ–ľ. –ź–≤—ā–ĺ—Ä—č –Ņ—Ä–Ķ–ī–Ņ–ĺ–Ľ–ĺ–∂–ł–Ľ–ł, —á—ā–ĺ GIST, –Ņ–ĺ –ļ—Ä–į–Ļ–Ĺ–Ķ–Ļ –ľ–Ķ—Ä–Ķ –Ķ–≥–ĺ –ľ–ł–ļ—Ä–ĺ—Ā–ļ–ĺ–Ņ–ł—á–Ķ—Ā–ļ–ł–Ķ —Ą–ĺ—Ä–ľ—č, —Ä–į—Ā–Ņ—Ä–ĺ—Ā—ā—Ä–į–Ĺ–Ķ–Ĺ—č –≥–ĺ—Ä–į–∑–ī–ĺ —ą–ł—Ä–Ķ, —á–Ķ–ľ –Ņ—Ä–ł–Ĺ—Ź—ā–ĺ –Ņ–ĺ–Ľ–į–≥–į—ā—Ć. –í–Ķ—Ä–ĺ—Ź—ā–Ĺ–ĺ, —á—ā–ĺ –≤ –Ī–ĺ–Ľ—Ć—ą–ł–Ĺ—Ā—ā–≤–Ķ —Ā–Ľ—É—á–į–Ķ–≤ –ĺ–Ĺ–ł –Ĺ–Ķ –ī–ĺ—Ā—ā–ł–≥–į—é—ā –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł –∑–Ĺ–į—á–ł–ľ—č—Ö —Ä–į–∑–ľ–Ķ—Ä–ĺ–≤ –ł –≤ —Ä—Ź–ī–Ķ —Ā–Ľ—É—á–į–Ķ–≤ —Ā–ļ–Ľ–ĺ–Ĺ–Ĺ—č –ļ —Ā–Ņ–ĺ–Ĺ—ā–į–Ĺ–Ĺ–ĺ–Ļ —Ä–Ķ–≥—Ä–Ķ—Ā—Ā–ł–ł.¬†
–ö—Ä–ĺ–ľ–Ķ —ā–ĺ–≥–ĺ, —Ā—ā–ĺ–ł—ā –Ņ–ĺ–ī—á–Ķ—Ä–ļ–Ĺ—É—ā—Ć, —á—ā–ĺ —ā–į–ļ–į—Ź –≤—č—Ā–ĺ–ļ–į—Ź —á–į—Ā—ā–ĺ—ā–į –≤—Ā—ā—Ä–Ķ—á–į–Ķ–ľ–ĺ—Ā—ā–ł –Ĺ–ł –≤ –ļ–ĺ–Ķ–Ļ –ľ–Ķ—Ä–Ķ –Ĺ–Ķ –ĺ–∑–Ĺ–į—á–į–Ķ—ā –ł—Ā—ā–ł–Ĺ–Ĺ–ĺ–≥–ĺ —Ä–į—Ā–Ņ—Ä–ĺ—Ā—ā—Ä–į–Ĺ–Ķ–Ĺ–ł—Ź GIST –∂–Ķ–Ľ—É–ī–ļ–į, –Ņ–ĺ—Ā–ļ–ĺ–Ľ—Ć–ļ—É –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ķ –Ņ—Ä–ĺ–≤–Ķ–ī–Ķ–Ĺ–ĺ –Ĺ–į –≤—č–Ī–ĺ—Ä–ļ–Ķ —Ä–į–ļ–į –∂–Ķ–Ľ—É–ī–ļ–į, –≥–ī–Ķ –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ—č –ļ–į–Ĺ—Ü–Ķ—Ä–ĺ–≥–Ķ–Ĺ–Ķ–∑–į –≤ –ĺ–Ņ—Ä–Ķ–ī–Ķ–Ľ–Ķ–Ĺ–Ĺ–ĺ–Ļ –ľ–Ķ—Ä–Ķ —Ā–į–ľ–ł –Ņ–ĺ —Ā–Ķ–Ī–Ķ –ľ–ĺ–≥—É—ā —Ā–Ņ–ĺ—Ā–ĺ–Ī—Ā—ā–≤–ĺ–≤–į—ā—Ć –Ņ–ĺ—Ź–≤–Ľ–Ķ–Ĺ–ł—é —Ā—ā—Ä–ĺ–ľ–į–Ľ—Ć–Ĺ—č—Ö –ĺ–Ņ—É—Ö–ĺ–Ľ–Ķ–Ļ. –í –°–Ķ–≤–Ķ—Ä–Ĺ–ĺ–Ļ –ź–ľ–Ķ—Ä–ł–ļ–Ķ —Ā–į–ľ–ĺ–Ķ –ļ—Ä—É–Ņ–Ĺ–ĺ–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ķ –Ņ–ĺ —ć–Ņ–ł–ī–Ķ–ľ–ł–ĺ–Ľ–ĺ–≥–ł–ł GIST –Ņ—Ä–ĺ–≤–Ķ–ī–Ķ–Ĺ–ĺ E.A. Perez –ł —Ā–ĺ–į–≤—ā. [17]. –í —Ä–į–Ī–ĺ—ā—É –Ī—č–Ľ–ł –≤–ļ–Ľ—é—á–Ķ–Ĺ—č –Ī–į–∑—č –ī–į–Ĺ–Ĺ—č—Ö —Ä–Ķ–≥–ł—Ā—ā—Ä–į SEER (Surveillance, Epidemiology and End Results database), –ļ–ĺ—ā–ĺ—Ä—č–Ļ –ĺ—Ö–≤–į—ā—č–≤–į–Ķ—ā –ĺ–ļ–ĺ–Ľ–ĺ 17% –Ĺ–į—Ā–Ķ–Ľ–Ķ–Ĺ–ł—Ź –°–®–ź, –ł –Ī–į–∑–į –ī–į–Ĺ–Ĺ—č—Ö –ĺ–Ĺ–ļ–ĺ–Ľ–ĺ–≥–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ —Ä–Ķ–≥–ł—Ā—ā—Ä–į –§–Ľ–ĺ—Ä–ł–ī—č (Florida Cancer Data System), –ĺ—Ö–≤–į—ā—č–≤–į—é—Č–į—Ź –ĺ–ļ–ĺ–Ľ–ĺ 6% –Ĺ–į—Ā–Ķ–Ľ–Ķ–Ĺ–ł—Ź –ź–ľ–Ķ—Ä–ł–ļ–ł. –Ē–į–Ĺ–Ĺ—č–Ķ SEER –ĺ–Ī—Ä–į–Ī–ĺ—ā–į–Ĺ—č —Ā 1992 –Ņ–ĺ 2005 –≥., —Ä–Ķ–≥–ł—Ā—ā—Ä–į –§–Ľ–ĺ—Ä–ł–ī—č ‚Äď —Ā 1984 –≥. –Ď—č–Ľ–ĺ —É—Ā—ā–į–Ĺ–ĺ–≤–Ľ–Ķ–Ĺ–ĺ, —á—ā–ĺ –≤ 1994 –≥. 93% –ĺ–Ņ—É—Ö–ĺ–Ľ–Ķ–Ļ —Ä–į—Ā—Ü–Ķ–Ĺ–ł–≤–į–Ľ–ł—Ā—Ć –ļ–į–ļ –≥–Ľ–į–ī–ļ–ĺ–ľ—č—ą–Ķ—á–Ĺ—č–Ķ –ĺ–Ņ—É—Ö–ĺ–Ľ–ł –ł —ā–ĺ–Ľ—Ć–ļ–ĺ 6% ‚Äď –ļ–į–ļ GIST. –í 2002 –≥. —É–∂–Ķ 82% —Ā–ĺ—Ā—ā–į–≤–Ľ—Ź–Ľ–ł GIST –ł —ā–ĺ–Ľ—Ć–ļ–ĺ 17% ‚Äď –≥–Ľ–į–ī–ļ–ĺ–ľ—č—ą–Ķ—á–Ĺ—č–Ķ –ĺ–Ņ—É—Ö–ĺ–Ľ–ł. –ü—Ä–ĺ–Ņ–ĺ—Ä—Ü–ł—Ź GIST –∂–Ķ–Ľ—É–ī–ļ–į –Ī—č–Ľ–į –Ķ—Č–Ķ –≤—č—ą–Ķ ‚Äď 96% –ī–Ľ—Ź SEER –ł 93% –ī–Ľ—Ź –Ņ–ĺ–Ņ—É–Ľ—Ź—Ü–ł–ł –§–Ľ–ĺ—Ä–ł–ī—č. –ó–į–Ī–ĺ–Ľ–Ķ–≤–į–Ķ–ľ–ĺ—Ā—ā—Ć GIST —É–≤–Ķ–Ľ–ł—á–ł–Ľ–į—Ā—Ć —Ā 0,028 –Ĺ–į 100 —ā—č—Ā. –Ĺ–į—Ā–Ķ–Ľ–Ķ–Ĺ–ł—Ź –≤¬†1992¬†–≥. –ī–ĺ 0,688 –Ĺ–į 100 —ā—č—Ā. –Ĺ–į—Ā–Ķ–Ľ–Ķ–Ĺ–ł—Ź –≤ 2005¬†–≥. –ź–≤—ā–ĺ—Ä—č –Ņ–ĺ–ī—á–Ķ—Ä–ļ–Ĺ—É–Ľ–ł, —á—ā–ĺ –Ņ–ĺ–≤—č—ą–Ķ–Ĺ–ł–Ķ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ķ–ľ–ĺ—Ā—ā–ł —Ā–≤—Ź–∑–į–Ĺ–ĺ –ļ–į–ļ —Ā —Ź–≤–Ĺ—č–ľ —É–Ľ—É—á—ą–Ķ–Ĺ–ł–Ķ–ľ –ī–ł–į–≥–Ĺ–ĺ—Ā—ā–ł–ļ–ł, —ā–į–ļ –ł —Ā —É–Ľ—É—á—ą–Ķ–Ĺ–ł–Ķ–ľ —Ā–ļ—Ä–ł–Ĺ–ł–Ĺ–≥–į. –ü–ĺ—Ā–Ľ–Ķ 2000 –≥. –ĺ–Ņ—É—Ö–ĺ–Ľ–ł –∂–Ķ–Ľ—É–ī–ļ–į —Ā–ĺ—Ā—ā–į–≤–Ľ—Ź–Ľ–ł 47% —Ā–Ľ—É—á–į–Ķ–≤, –ĺ–Ņ—É—Ö–ĺ–Ľ–ł —ā–ĺ–Ĺ–ļ–ĺ–Ļ –ļ–ł—ą–ļ–ł ‚Äď 34%. –ė–Ĺ—ā–Ķ—Ä–Ķ—Ā–Ĺ–ĺ, —á—ā–ĺ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ķ–ľ–ĺ—Ā—ā—Ć GIST —ɬ†–į—Ą—Ä–ĺ–į–ľ–Ķ—Ä–ł–ļ–į–Ĺ—Ü–Ķ–≤ –≤—č—ą–Ķ, —á–Ķ–ľ —ɬ†–Ī–Ķ–Ľ–ĺ–≥–ĺ –Ĺ–į—Ā–Ķ–Ľ–Ķ–Ĺ–ł—Ź.
–Ę–į—Ä–≥–Ķ—ā–Ĺ–į—Ź —ā–Ķ—Ä–į–Ņ–ł—Ź –ľ–Ķ—ā–į—Ā—ā–į—ā–ł—á–Ķ—Ā–ļ–ł—Ö —Ą–ĺ—Ä–ľ GIST
–ď–Ľ–ł–≤–Ķ–ļ –Ĺ–į—á–į–Ľ –Ņ—Ä–ł–ľ–Ķ–Ĺ—Ź—ā—Ć—Ā—Ź –≤ 90-–Ķ –≥–ĺ–ī—č –Ņ—Ä–ĺ—ą–Ľ–ĺ–≥–ĺ —Ā—ā–ĺ–Ľ–Ķ—ā–ł—Ź –≤ –ļ–į—á–Ķ—Ā—ā–≤–Ķ –ł–Ĺ–≥–ł–Ī–ł—ā–ĺ—Ä–į —ā–ł—Ä–ĺ–∑–ł–Ĺ–ļ–ł–Ĺ–į–∑ –ī–Ľ—Ź –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź —Ö—Ä–ĺ–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –ľ–ł–Ķ–Ľ–ĺ–Ľ–Ķ–Ļ–ļ–ĺ–∑–į –Ī–Ľ–į–≥–ĺ–ī–į—Ä—Ź —Ā–Ņ–ĺ—Ā–ĺ–Ī–Ĺ–ĺ—Ā—ā–ł –ł–Ĺ–≥–ł–Ī–ł—Ä–ĺ–≤–į—ā—Ć –ĺ–Ĺ–ļ–ĺ–Ņ—Ä–ĺ—ā–Ķ–ł–Ĺ BCR-ABL. –í–Ņ–ĺ—Ā–Ľ–Ķ–ī—Ā—ā–≤–ł–ł –Ĺ–į–Ī–Ľ—é–ī–Ķ–Ĺ–ł–Ķ, —á—ā–ĺ –Ī–Ķ–Ľ–ļ–ł BCR-ABL –ł KIT –ł–ľ–Ķ—é—ā —Ā—ā—Ä—É–ļ—ā—É—Ä–Ĺ—É—é —Ā—Ö–ĺ–∂–Ķ—Ā—ā—Ć, –ł–Ĺ—Ā–Ņ–ł—Ä–ł—Ä–ĺ–≤–į–Ľ–ĺ –Ņ—Ä–ĺ–≤–Ķ–ī–Ķ–Ĺ–ł–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ –Ņ–ĺ –ł—Ā–Ņ–ĺ–Ľ—Ć–∑–ĺ–≤–į–Ĺ–ł—é –ď–Ľ–ł–≤–Ķ–ļ–į –Ĺ–į –ļ–Ľ–Ķ—ā–ĺ—á–Ĺ—č—Ö –Ľ–ł–Ĺ–ł—Ź—Ö, –≤ —ā–ĺ–ľ —á–ł—Ā–Ľ–Ķ –ł –Ĺ–į –ļ–Ľ–Ķ—ā–ļ–į—Ö GIST [19]. –ü—Ä–į–ļ—ā–ł—á–Ķ—Ā–ļ–ł —Ā—Ä–į–∑—É –∂–Ķ –Ī—č–Ľ–ł –ł–Ĺ–ł—Ü–ł–ł—Ä–ĺ–≤–į–Ĺ—č –Ņ—Ä–ĺ—ā–ĺ–ļ–ĺ–Ľ—č –Ņ–ĺ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—é —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā–ł –ī–≤–ĺ–Ļ–Ĺ–ĺ–Ļ –ī–ĺ–∑—č –ď–Ľ–ł–≤–Ķ–ļ–į –Ņ—Ä–ł –ľ–Ķ—ā–į—Ā—ā–į—ā–ł—á–Ķ—Ā–ļ–ł—Ö —Ą–ĺ—Ä–ľ–į—Ö –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź. –í —Ü–Ķ–Ľ–ĺ–ľ –ď–Ľ–ł–≤–Ķ–ļ –Ī—č–Ľ —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ķ–Ĺ —É 70‚Äď85% –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤, –ľ–Ķ–ī–ł–į–Ĺ–į –≤—Ä–Ķ–ľ–Ķ–Ĺ–ł –ī–ĺ –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź —Ā–ĺ—Ā—ā–į–≤–ł–Ľ–į 20‚Äď24 –ľ–Ķ—Ā. [20].
–í—č—Ā–ĺ–ļ–į—Ź —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć –ď–Ľ–ł–≤–Ķ–ļ–į –Ņ–ĺ—Ä–ĺ–ī–ł–Ľ–į —É–Ī–Ķ–∂–ī–Ķ–Ĺ–ł–Ķ, —á—ā–ĺ –≤–ĺ–∑–ľ–ĺ–∂–Ĺ–ĺ –Ņ–ĺ–Ľ–Ĺ–ĺ–Ķ –ł–∑–Ľ–Ķ—á–Ķ–Ĺ–ł–Ķ –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö —Ā GIST: –≤–ĺ–∑–Ĺ–ł–ļ–Ľ–ĺ –∂–Ķ–Ľ–į–Ĺ–ł–Ķ –Ņ—Ä–Ķ–ļ—Ä–į—ā–ł—ā—Ć –Ņ—Ä–ł–Ķ–ľ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į –Ņ–ĺ—Ā–Ľ–Ķ —É—Ā–Ņ–Ķ—ą–Ĺ–ĺ–Ļ –ī–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ļ —Ä–Ķ–ľ–ł—Ā—Ā–ł–ł. –í 2002 –≥. —Ą—Ä–į–Ĺ—Ü—É–∑—Ā–ļ–ł–ľ–ł –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į—ā–Ķ–Ľ—Ź–ľ–ł –ł–Ĺ–ł—Ü–ł–ł—Ä–ĺ–≤–į–Ĺ–ĺ —Ä–į–Ĺ–ī–ĺ–ľ–ł–∑–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ķ III —Ą–į–∑—č BFR14, –ļ–ĺ—ā–ĺ—Ä–ĺ–Ķ –Ņ–ĺ—Ā—ā–į–≤–ł–Ľ–ĺ –≤–ĺ–Ņ—Ä–ĺ—Ā –ĺ –Ņ—Ä–ĺ–ī–ĺ–Ľ–∂–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ—Ā—ā–ł –Ņ—Ä–ł–Ķ–ľ–į –ď–Ľ–ł–≤–Ķ–ļ–į —É –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö —Ā –ľ–Ķ—ā–į—Ā—ā–į—ā–ł—á–Ķ—Ā–ļ–ĺ–Ļ —Ą–ĺ—Ä–ľ–ĺ–Ļ –Ī–ĺ–Ľ–Ķ–∑–Ĺ–ł. –Ď–ĺ–Ľ—Ć–Ĺ—č–Ķ –Ī—č–Ľ–ł —Ä–į–Ĺ–ī–ĺ–ľ–ł–∑–ł—Ä–ĺ–≤–į–Ĺ—č –Ĺ–į –≥—Ä—É–Ņ–Ņ—č –Ņ–ĺ—Ā—ā–ĺ—Ź–Ĺ–Ĺ–ĺ–≥–ĺ –Ņ—Ä–ł–Ķ–ľ–į –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į (C-group, continuous) –ł –ĺ–ļ–ĺ–Ĺ—á–į–Ĺ–ł—Ź –Ņ—Ä–ł–Ķ–ľ–į —á–Ķ—Ä–Ķ–∑ –≥–ĺ–ī —É—Ā–Ņ–Ķ—ą–Ĺ–ĺ–≥–ĺ –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź –ď–Ľ–ł–≤–Ķ–ļ–ĺ–ľ (S-group, stop). –Ē–ł–∑–į–Ļ–Ĺ –Ņ–ĺ–ī—Ä–į–∑—É–ľ–Ķ–≤–į–Ľ –≤–ĺ–∑–ĺ–Ī–Ĺ–ĺ–≤–Ľ–Ķ–Ĺ–ł–Ķ –Ņ—Ä–ł–Ķ–ľ–į –ď–Ľ–ł–≤–Ķ–ļ–į –≤ S-–≥—Ä—É–Ņ–Ņ–Ķ –≤ —Ā–Ľ—É—á–į–Ķ –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź. –í –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ķ –Ī—č–Ľ–ĺ –≤–ļ–Ľ—é—á–Ķ–Ĺ–ĺ 58 –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤. –í –Ņ–ĺ—Ā–Ľ–Ķ–ī—É—é—Č–Ķ–ľ —ć—ā–ĺ—ā –Ņ—Ä–ĺ—ā–ĺ–ļ–ĺ–Ľ —ā—Ä–į–Ĺ—Ā—Ą–ĺ—Ä–ľ–ł—Ä–ĺ–≤–į–Ľ—Ā—Ź –≤ —Ä–į–Ĺ–ī–ĺ–ľ–ł–∑–į—Ü–ł—é –°- –ł S-–≥—Ä—É–Ņ–Ņ —á–Ķ—Ä–Ķ–∑ 3 –≥–ĺ–ī–į (50 –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤) –ł —á–Ķ—Ä–Ķ–∑ 5 –Ľ–Ķ—ā (25 –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤). –í stop-–≥—Ä—É–Ņ–Ņ–Ķ –ļ–į–∂–ī—č–Ļ —Ä–į–∑ –Ņ–ĺ—Ā–Ľ–Ķ –ĺ–ļ–ĺ–Ĺ—á–į–Ĺ–ł—Ź –Ņ—Ä–ł–Ķ–ľ–į –ď–Ľ–ł–≤–Ķ–ļ–į –ī–ĺ—Ā—ā–ĺ–≤–Ķ—Ä–Ĺ–ĺ –Ī—č—Ā—ā—Ä–Ķ–Ķ –Ĺ–į—Ā—ā—É–Ņ–į–Ľ–ĺ –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ [25‚Äď27]. –Ę–į–ļ, —á–Ķ—Ä–Ķ–∑ 5 –Ľ–Ķ—ā –Ņ–ĺ—Ā–Ľ–Ķ —Ä–į–Ĺ–ī–ĺ–ľ–ł–∑–į—Ü–ł–ł –Ĺ–ł —É –ĺ–ī–Ĺ–ĺ–≥–ĺ –ł–∑ 12 –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤, –≤–ĺ—ą–Ķ–ī—ą–ł—Ö –≤ –°-–≥—Ä—É–Ņ–Ņ—É, –Ĺ–Ķ –ĺ—ā–ľ–Ķ—á–Ķ–Ĺ–ĺ —Ä–Ķ—Ü–ł–ī–ł–≤–ĺ–≤, –≤ S-–≥—Ä—É–Ņ–Ņ–Ķ ‚Äď 7 —Ä–Ķ—Ü–ł–ī–ł–≤–ĺ–≤ –ł–∑ 20 (—Ä = 0,0317). –ź–≤—ā–ĺ—Ä—č —Ā–ī–Ķ–Ľ–į–Ľ–ł –≤—č–≤–ĺ–ī, —á—ā–ĺ –Ņ–į—Ü–ł–Ķ–Ĺ—ā—č —Ā –ľ–Ķ—ā–į—Ā—ā–į—ā–ł—á–Ķ—Ā–ļ–ĺ–Ļ –Ī–ĺ–Ľ–Ķ–∑–Ĺ—Ć—é –ī–ĺ–Ľ–∂–Ĺ—č –Ņ–ĺ–Ľ—É—á–į—ā—Ć –ď–Ľ–ł–≤–Ķ–ļ –Ņ–ĺ—Ā—ā–ĺ—Ź–Ĺ–Ĺ–ĺ –ī–ĺ –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź –Ľ–ł–Ī–ĺ –ī–ĺ –Ĺ–į—Ā—ā—É–Ņ–Ľ–Ķ–Ĺ–ł—Ź –Ĺ–Ķ–Ņ–Ķ—Ä–Ķ–Ĺ–ĺ—Ā–ł–ľ–ĺ—Ā—ā–ł –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į. –Ē–ĺ—Ā—ā–ĺ–≤–Ķ—Ä–Ĺ—č—Ö —Ä–į–∑–Ľ–ł—á–ł–Ļ –≤ –ĺ–Ī—Č–Ķ–Ļ –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā–ł –ľ–Ķ–∂–ī—É –ī–≤—É–ľ—Ź –≥—Ä—É–Ņ–Ņ–į–ľ–ł –Ĺ–Ķ –Ī—č–Ľ–ĺ. –Ę–į–ļ–ł–ľ –ĺ–Ī—Ä–į–∑–ĺ–ľ, –ĺ–ļ–ĺ–Ĺ—á–į–Ĺ–ł–Ķ –Ņ—Ä–ł–Ķ–ľ–į –Ľ–Ķ–ļ–į—Ä—Ā—ā–≤–į –ī–į–∂–Ķ –Ņ–ĺ—Ā–Ľ–Ķ –ī–Ľ–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ–≥–ĺ —É—Ā–Ņ–Ķ—ą–Ĺ–ĺ–≥–ĺ –Ņ—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł—Ź –į—Ā—Ā–ĺ—Ü–ł–ł—Ä—É–Ķ—ā—Ā—Ź —Ā –Ī—č—Ā—ā—Ä—č–ľ –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ–ľ –Ī–ĺ–Ľ–Ķ–∑–Ĺ–ł.
–Ě–Ķ–ĺ- –ł –į–ī—ä—é–≤–į–Ĺ—ā–Ĺ–į—Ź —ā–Ķ—Ä–į–Ņ–ł—Ź GIST
–ü–Ķ—Ä–≤—č–Ļ –ĺ–Ņ—č—ā –Ņ—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł—Ź –ď–Ľ–ł–≤–Ķ–ļ–į –Ņ—Ä–ł GIST –≤–ĺ –≤—Ā–Ķ—Ö —Ā—ā—Ä–į–Ĺ–į—Ö –Ņ–ĺ–ļ–į–∑–į–Ľ –≤—č—Ā–ĺ–ļ—É—é —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į. –ü–Ķ—Ä–≤—č–Ķ –Ņ—Ä–ĺ—ā–ĺ–ļ–ĺ–Ľ—č –ł–∑—É—á–į–Ľ–ł –ľ–Ķ—ā–į—Ā—ā–į—ā–ł—á–Ķ—Ā–ļ–ł–Ķ —Ą–ĺ—Ä–ľ—č –Ī–ĺ–Ľ–Ķ–∑–Ĺ–ł, –∑–į—ā–Ķ–ľ –Ņ–ĺ—á—ā–ł –ĺ–ī–Ĺ–ĺ–≤—Ä–Ķ–ľ–Ķ–Ĺ–Ĺ–ĺ –Ī—č–Ľ–ł –ł–Ĺ–ł—Ü–ł–ł—Ä–ĺ–≤–į–Ĺ—č –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź –Ņ–ĺ –į–ī—ä—é–≤–į–Ĺ—ā–Ĺ–ĺ–Ļ –ł –Ĺ–Ķ–ĺ–į–ī—ä—é–≤–į–Ĺ—ā–Ĺ–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł. –ö 2011 –≥. –Ī—č–Ľ–ł –Ĺ–į–ļ–ĺ–Ņ–Ľ–Ķ–Ĺ—č –ī–į–Ĺ–Ĺ—č–Ķ, –Ņ–ĺ–ļ–į–∑–į–≤—ą–ł–Ķ 25%-–Ĺ—É—é –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā—Ć –∑–į 9 –Ľ–Ķ—ā –Ņ—Ä–ł –ľ–Ķ—ā–į—Ā—ā–į—ā–ł—á–Ķ—Ā–ļ–ĺ–Ļ —Ą–ĺ—Ä–ľ–Ķ –Ī–ĺ–Ľ–Ķ–∑–Ĺ–ł [1] –ł —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć —ā—Ä–Ķ—Ö–Ľ–Ķ—ā–Ĺ–Ķ–Ļ –į–ī—ä—é–≤–į–Ĺ—ā–Ĺ–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł –Ņ–ĺ—Ā–Ľ–Ķ —Ä–į–ī–ł–ļ–į–Ľ—Ć–Ĺ—č—Ö –ĺ–Ņ–Ķ—Ä–į—Ü–ł–Ļ [28, 29].
–ě—Ā–Ĺ–ĺ–≤–Ĺ–ĺ–Ļ —Ü–Ķ–Ľ—Ć—é –Ņ—Ä–Ķ–ī–ĺ–Ņ–Ķ—Ä–į—Ü–ł–ĺ–Ĺ–Ĺ–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł –Ī–ĺ–Ľ—Ć—ą–ł–Ĺ—Ā—ā–≤–ĺ –į–≤—ā–ĺ—Ä–ĺ–≤ —Ā—á–ł—ā–į–Ľ–ł —É–ľ–Ķ–Ĺ—Ć—ą–Ķ–Ĺ–ł–Ķ —Ä–į–∑–ľ–Ķ—Ä–ĺ–≤ –ĺ–Ņ—É—Ö–ĺ–Ľ–ł. –í —á–į—Ā—ā–Ĺ–ĺ—Ā—ā–ł, —ā–Ķ–ĺ—Ä–Ķ—ā–ł—á–Ķ—Ā–ļ–ł –ĺ–Ņ—Ä–į–≤–ī–į–Ĺ–į –Ĺ–Ķ–ĺ–į–ī—ä—é–≤–į–Ĺ—ā–Ĺ–į—Ź —ā–Ķ—Ä–į–Ņ–ł—Ź –Ņ—Ä–ł –Ľ–ĺ–ļ–į–Ľ–ł–∑–į—Ü–ł–ł GIST –≤ –ī–≤–Ķ–Ĺ–į–ī—Ü–į—ā–ł–Ņ–Ķ—Ä—Ā—ā–Ĺ–ĺ–Ļ –ł –Ņ—Ä—Ź–ľ–ĺ–Ļ –ļ–ł—ą–ļ–Ķ. –†–Ķ—ā—Ä–ĺ—Ā–Ņ–Ķ–ļ—ā–ł–≤–Ĺ—č–Ļ –į–Ĺ–į–Ľ–ł–∑ 46 –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā –Ņ–Ķ—Ä–≤–ł—á–Ĺ—č–ľ–ł –ł —Ä–Ķ—Ü–ł–ī–ł–≤–Ĺ—č–ľ–ł –ĺ–Ņ—É—Ö–ĺ–Ľ—Ź–ľ–ł –Ņ–ĺ–ļ–į–∑–į–Ľ, —á—ā–ĺ –Ņ—Ä–ł –Ņ—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł–ł –ď–Ľ–ł–≤–Ķ–ļ–į –≤ –Ņ—Ä–Ķ–ī–ĺ–Ņ–Ķ—Ä–į—Ü–ł–ĺ–Ĺ–Ĺ–ĺ–ľ —Ä–Ķ–∂–ł–ľ–Ķ —É–≤–Ķ–Ľ–ł—á–ł–≤–į–Ľ—Ā—Ź —ą–į–Ĺ—Ā –Ĺ–į –Ņ–ĺ–Ľ–Ĺ–ĺ–Ķ —É–ī–į–Ľ–Ķ–Ĺ–ł–Ķ –ĺ–Ņ—É—Ö–ĺ–Ľ–ł [30]. –í —Ä–į–ľ–ļ–į—Ö –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź –Ņ–į—Ü–ł–Ķ–Ĺ—ā–į–ľ –≤—č–Ņ–ĺ–Ľ–Ĺ—Ź–Ľ–ł –ļ–ĺ–ľ–Ņ—Ć—é—ā–Ķ—Ä–Ĺ—É—é —ā–ĺ–ľ–ĺ–≥—Ä–į—Ą–ł—é –ļ–į–∂–ī—č–Ķ 2‚Äď3 –ľ–Ķ—Ā. –ī–Ľ—Ź –ĺ—Ü–Ķ–Ĺ–ļ–ł —Ā—ā–Ķ–Ņ–Ķ–Ĺ–ł —É–ľ–Ķ–Ĺ—Ć—ą–Ķ–Ĺ–ł—Ź –ĺ–Ņ—É—Ö–ĺ–Ľ–ł. –ě–ī–ł–Ĺ–Ĺ–į–ī—Ü–į—ā—Ć –ł–∑ 46 –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö –Ņ–ĺ–Ľ—É—á–į–Ľ–ł —ā–Ķ—Ä–į–Ņ–ł—é –ī–ĺ –ĺ–Ņ–Ķ—Ä–į—Ü–ł–ł —Ā –ľ–Ķ–ī–ł–į–Ĺ–ĺ–Ļ 11,9 –ľ–Ķ—Ā. –ü—Ä–ł –ľ–Ķ–ī–ł–į–Ĺ–Ķ –Ĺ–į–Ī–Ľ—é–ī–Ķ–Ĺ–ł—Ź 19,5 –ľ–Ķ—Ā. —É 11 –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö –Ĺ–Ķ –Ĺ–į–Ī–Ľ—é–ī–į–Ľ–ĺ—Ā—Ć –Ņ—Ä–ł–∑–Ĺ–į–ļ–ĺ–≤ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź. –í –ī—Ä—É–≥–ĺ–ľ –Ņ—Ä–ĺ—Ā–Ņ–Ķ–ļ—ā–ł–≤–Ĺ–ĺ–ľ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–ł –ł–∑ 36 –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —É 33 –Ņ—Ä–ł –ľ–Ķ–ī–ł–į–Ĺ–Ķ 11 –ľ–Ķ—Ā. –≤ –Ņ—Ä–Ķ–ī–ĺ–Ņ–Ķ—Ä–į—Ü–ł–ĺ–Ĺ–Ĺ–ĺ–ľ —Ä–Ķ–∂–ł–ľ–Ķ —Ä–į–∑–ľ–Ķ—Ä –ĺ–Ņ—É—Ö–ĺ–Ľ–ł –≤ —Ā—Ä–Ķ–ī–Ĺ–Ķ–ľ —É–ľ–Ķ–Ĺ—Ć—ą–ł–Ľ—Ā—Ź —Ā 10,5 –ī–ĺ 5,5 —Ā–ľ. –ü—Ä–ł —ć—ā–ĺ–ľ 83% –ł–∑–Ĺ–į—á–į–Ľ—Ć–Ĺ–ĺ –Ĺ–Ķ–ĺ–Ņ–Ķ—Ä–į–Ī–Ķ–Ľ—Ć–Ĺ—č—Ö –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā—ā–į–Ľ–ł –ĺ–Ņ–Ķ—Ä–į–Ī–Ķ–Ľ—Ć–Ĺ—č–ľ–ł [31]. –ě—á–Ķ–≤–ł–ī–Ĺ–ĺ, —á—ā–ĺ —Ü–Ķ–Ľ–Ķ—Ā–ĺ–ĺ–Ī—Ä–į–∑–Ĺ–ĺ—Ā—ā—Ć –Ņ—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł—Ź –ď–Ľ–ł–≤–Ķ–ļ–į –ī–ĺ –ĺ–Ņ–Ķ—Ä–į—Ü–ł–ł –ī–ĺ–Ľ–∂–Ĺ–į –ĺ—Ü–Ķ–Ĺ–ł–≤–į—ā—Ć—Ā—Ź –≤–ľ–Ķ—Ā—ā–Ķ —Ā —Ö–ł—Ä—É—Ä–≥–ĺ–ľ –ł –ī–ĺ–Ľ–∂–Ĺ–į —É—á–ł—ā—č–≤–į—ā—Ć—Ā—Ź –≤–ĺ–∑–ľ–ĺ–∂–Ĺ–ĺ—Ā—ā—Ć —É–ľ–Ķ–Ĺ—Ć—ą–ł—ā—Ć —Ä–į–∑–ľ–Ķ—Ä—č –ĺ–Ņ—É—Ö–ĺ–Ľ–ł, –ĺ—Ā–ĺ–Ī–Ķ–Ĺ–Ĺ–ĺ –Ņ—Ä–ł –Ķ–Ķ –Ľ–ĺ–ļ–į–Ľ–ł–∑–į—Ü–ł–ł –≤ –Ĺ–Ķ–Ņ–ĺ—Ā—Ä–Ķ–ī—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ–Ļ –Ī–Ľ–ł–∑–ĺ—Ā—ā–ł –ļ –ļ–į—Ä–ī–ł–ł, –≤ –ī–≤–Ķ–Ĺ–į–ī—Ü–į—ā–ł–Ņ–Ķ—Ä—Ā—ā–Ĺ–ĺ–Ļ –ł –Ņ—Ä—Ź–ľ–ĺ–Ļ –ļ–ł—ą–ļ–Ķ.
–≠—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć –Ņ—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł—Ź —ā–į—Ä–≥–Ķ—ā–Ĺ–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł –Ņ—Ä–ł –ľ–Ķ—ā–į—Ā—ā–į—ā–ł—á–Ķ—Ā–ļ–ł—Ö —Ą–ĺ—Ä–ľ–į—Ö GIST –Ĺ–Ķ –ľ–ĺ–≥–Ľ–į –Ĺ–Ķ –ł–Ĺ–ł—Ü–ł–ł—Ä–ĺ–≤–į—ā—Ć –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź –Ņ–ĺ –į–ī—ä—é–≤–į–Ĺ—ā–Ĺ–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł GIST. –ě–ī–Ĺ–ĺ –ł–∑ —Ā–į–ľ—č—Ö –ļ—Ä—É–Ņ–Ĺ—č—Ö —Ä–į–Ĺ–ī–ĺ–ľ–ł–∑–ł—Ä–ĺ–≤–į–Ĺ–Ĺ—č—Ö (–Ņ–Ľ–į—Ü–Ķ–Ī–ĺ–ļ–ĺ–Ĺ—ā—Ä–ĺ–Ľ–ł—Ä—É–Ķ–ľ–ĺ–Ķ, –ī–≤–ĺ–Ļ–Ĺ–ĺ–Ķ —Ā–Ľ–Ķ–Ņ–ĺ–Ķ) –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–Ļ –Ņ–ĺ –į–ī—ä—é–≤–į–Ĺ—ā–Ĺ–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł GIST (ACOSOG Z9001) –Ī—č–Ľ–ĺ –Ņ—Ä–ĺ–≤–Ķ–ī–Ķ–Ĺ–ĺ –≤ –°–®–ź. –° 2002 –Ņ–ĺ 2007 –≥. –≤ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–ł —É—á–į—Ā—ā–≤–ĺ–≤–į–Ľ–ł 230 –ļ–Ľ–ł–Ĺ–ł–ļ –°–®–ź –ł –ö–į–Ĺ–į–ī—č. –ö—Ä–ł—ā–Ķ—Ä–ł—Ź–ľ–ł –≤–ļ–Ľ—é—á–Ķ–Ĺ–ł—Ź –Ī—č–Ľ–ł —Ā–Ľ–Ķ–ī—É—é—Č–ł–Ķ –Ņ–ĺ–ļ–į–∑–į—ā–Ķ–Ľ–ł: –ĺ–Ņ—É—Ö–ĺ–Ľ—Ć –Ī–ĺ–Ľ–Ķ–Ķ 3 —Ā–ľ –≤ –ī–ł–į–ľ–Ķ—ā—Ä–Ķ, —Ä–į–ī–ł–ļ–į–Ľ—Ć–Ĺ–į—Ź (–Ľ–ł–Ī–ĺ R1) –ĺ–Ņ–Ķ—Ä–į—Ü–ł—Ź, –≤–ĺ–∑—Ä–į—Ā—ā –Ņ–į—Ü–ł–Ķ–Ĺ—ā–į –Ī–ĺ–Ľ–Ķ–Ķ 18 –Ľ–Ķ—ā, CD117-–Ņ–ĺ–Ľ–ĺ–∂–ł—ā–Ķ–Ľ—Ć–Ĺ–į—Ź –ĺ–Ņ—É—Ö–ĺ–Ľ—Ć. –ü—Ä–ł –≤–ĺ–∑–Ĺ–ł–ļ–Ĺ–ĺ–≤–Ķ–Ĺ–ł–ł —Ä–Ķ—Ü–ł–ī–ł–≤–į –Ī–ĺ–Ľ–Ķ–∑–Ĺ–ł ¬ę–ĺ—Ā–Ľ–Ķ–Ņ–Ľ–Ķ–Ĺ–ł–Ķ¬Ľ –Ņ—Ä–Ķ–ļ—Ä–į—Č–į–Ľ–ĺ—Ā—Ć –ł –Ī–ĺ–Ľ—Ć–Ĺ—č–ľ –ł–∑ –Ņ–Ľ–į—Ü–Ķ–Ī–ĺ-–≥—Ä—É–Ņ–Ņ—č –Ņ—Ä–Ķ–ī–Ľ–į–≥–į–Ľ–ĺ—Ā—Ć –Ľ–Ķ—á–Ķ–Ĺ–ł–Ķ –ď–Ľ–ł–≤–Ķ–ļ–ĺ–ľ. –†–į–Ĺ–ī–ĺ–ľ–ł–∑–į—Ü–ł—Ź –Ī—č–Ľ–į –Ņ—Ä–ĺ–≤–Ķ–ī–Ķ–Ĺ–į —É 713 –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤.¬†
–í –≥—Ä—É–Ņ–Ņ–Ķ –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź –ď–Ľ–ł–≤–Ķ–ļ–ĺ–ľ –Ĺ–į–Ī–Ľ—é–ī–į–Ľ–ĺ—Ā—Ć 30 —Ā–ĺ–Ī—č—ā–ł–Ļ –ł 5 –Ľ–Ķ—ā–į–Ľ—Ć–Ĺ—č—Ö –ł—Ā—Ö–ĺ–ī–ĺ–≤ –ĺ—ā —Ä–į–∑–Ĺ—č—Ö –Ņ—Ä–ł—á–ł–Ĺ –Ī–Ķ–∑ —Ä–Ķ—Ü–ł–ī–ł–≤–į. –í –≥—Ä—É–Ņ–Ņ–Ķ –Ņ–Ľ–į—Ü–Ķ–Ī–ĺ –∑–į—Ą–ł–ļ—Ā–ł—Ä–ĺ–≤–į–Ĺ–ĺ 70 —Ā–ĺ–Ī—č—ā–ł–Ļ, –Ņ—Ä–ł —ć—ā–ĺ–ľ 69 –≤–ĺ–∑–≤—Ä–į—ā–ĺ–≤ –Ī–ĺ–Ľ–Ķ–∑–Ĺ–ł, 1 –Ľ–Ķ—ā–į–Ľ—Ć–Ĺ—č–Ļ –ł—Ā—Ö–ĺ–ī –Ī–Ķ–∑ —Ä–Ķ—Ü–ł–ī–ł–≤–į, 7 –Ľ–Ķ—ā–į–Ľ—Ć–Ĺ—č—Ö –ł—Ā—Ö–ĺ–ī–ĺ–≤ –Ņ–ĺ—Ā–Ľ–Ķ —Ä–į–∑–≤–ł—ā–ł—Ź —Ä–Ķ—Ü–ł–ī–ł–≤–į –Ī–ĺ–Ľ–Ķ–∑–Ĺ–ł, 5 ‚Äď –ĺ—ā –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź GIST, 2 ‚Äď –ĺ—ā –ī—Ä—É–≥–ł—Ö –Ņ—Ä–ł—á–ł–Ĺ. –Ě–į–ī–ĺ –ĺ—ā–ľ–Ķ—ā–ł—ā—Ć, —á—ā–ĺ –Ņ–Ķ—Ä–≤—č–Ķ –Ņ—Ä–ĺ–ľ–Ķ–∂—É—ā–ĺ—á–Ĺ—č–Ķ —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā—č —ć—ā–ĺ–≥–ĺ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź –Ī—č–Ľ–ł –ĺ–Ņ—É–Ī–Ľ–ł–ļ–ĺ–≤–į–Ĺ—č –≤ 2007 –≥. –ü—Ä–ł —ć—ā–ĺ–ľ –Ī—č–Ľ–ĺ —É—Ā—ā–į–Ĺ–ĺ–≤–Ľ–Ķ–Ĺ–ĺ, —á—ā–ĺ –į–ī—ä—é–≤–į–Ĺ—ā–Ĺ–į—Ź —ā–Ķ—Ä–į–Ņ–ł—Ź –ď–Ľ–ł–≤–Ķ–ļ–ĺ–ľ –≤ —ā–Ķ—á–Ķ–Ĺ–ł–Ķ –≥–ĺ–ī–į —Ā–Ņ–ĺ—Ā–ĺ–Ī—Ā—ā–≤–ĺ–≤–į–Ľ–į —É–Ľ—É—á—ą–Ķ–Ĺ–ł—é –Ņ–ĺ–ļ–į–∑–į—ā–Ķ–Ľ–Ķ–Ļ –Ī–Ķ–∑—Ä–Ķ—Ü–ł–ī–ł–≤–Ĺ–ĺ–Ļ –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā–ł —Ā 83% –≤ –ļ–ĺ–Ĺ—ā—Ä–ĺ–Ľ—Ć–Ĺ–ĺ–Ļ –≥—Ä—É–Ņ–Ņ–Ķ –ī–ĺ 97% –≤ –≥—Ä—É–Ņ–Ņ–Ķ –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź. –ě—ā—Ā—É—ā—Ā—ā–≤–ł–Ķ —Ä–į–∑–Ľ–ł—á–ł–Ļ –≤ –ĺ–Ī—Č–Ķ–Ļ –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā–ł –ĺ–Ī—ä—Ź—Ā–Ĺ—Ź–Ľ–ĺ—Ā—Ć –ļ–ĺ—Ä–ĺ—ā–ļ–ł–ľ —Ā—Ä–ĺ–ļ–ĺ–ľ –Ĺ–į–Ī–Ľ—é–ī–Ķ–Ĺ–ł—Ź –ł –Ĺ–į–∑–Ĺ–į—á–Ķ–Ĺ–ł–Ķ–ľ —ā–Ķ—Ä–į–Ņ–ł–ł –ď–Ľ–ł–≤–Ķ–ļ–ĺ–ľ –≤ —Ā–Ľ—É—á–į–Ķ —Ä–į–∑–≤–ł—ā–ł—Ź —Ä–Ķ—Ü–ł–ī–ł–≤–į –Ī–ĺ–Ľ–Ķ–∑–Ĺ–ł. –í 2009¬†–≥. –ĺ–Ņ—É–Ī–Ľ–ł–ļ–ĺ–≤–į–Ĺ—č —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā—č —ā—Ä–Ķ—Ö–Ľ–Ķ—ā–Ĺ–Ķ–≥–ĺ –Ĺ–į–Ī–Ľ—é–ī–Ķ–Ĺ–ł—Ź –∑–į –Ī–ĺ–Ľ—Ć–Ĺ—č–ľ–ł [32]. –ė–Ĺ—ā–Ķ—Ä–Ķ—Ā–Ĺ–ĺ, —á—ā–ĺ –ī–į–∂–Ķ –≤ –≥—Ä—É–Ņ–Ņ–Ķ –Ĺ–ł–∑–ļ–ĺ–≥–ĺ —Ä–ł—Ā–ļ–į —Ä–Ķ—Ü–ł–ī–ł–≤–į –Ī—č–Ľ–ł –∑–į—Ą–ł–ļ—Ā–ł—Ä–ĺ–≤–į–Ĺ—č —Ā–Ľ—É—á–į–ł —Ä–Ķ—Ü–ł–ī–ł–≤–ĺ–≤ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź.¬†
–í –≥—Ä—É–Ņ–Ņ–Ķ –≤—č—Ā–ĺ–ļ–ĺ–≥–ĺ —Ä–ł—Ā–ļ–į —Ä–Ķ—Ü–ł–ī–ł–≤–į (–ĺ–Ņ—É—Ö–ĺ–Ľ–ł –Ī–ĺ–Ľ–Ķ–Ķ 10 —Ā–ľ –≤ –ī–ł–į–ľ–Ķ—ā—Ä–Ķ) –į–ī—ä—é–≤–į–Ĺ—ā–Ĺ–į—Ź —ā–Ķ—Ä–į–Ņ–ł—Ź –Ņ–ĺ–ļ–į–∑–į–Ľ–į –Ĺ–į–ł–Ī–ĺ–Ľ–Ķ–Ķ –≤—č—Ā–ĺ–ļ—É—é —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć, —ā–Ķ–ľ –Ĺ–Ķ –ľ–Ķ–Ĺ–Ķ–Ķ –Ņ–ĺ—Ā–Ľ–Ķ –ĺ–ļ–ĺ–Ĺ—á–į–Ĺ–ł—Ź –Ņ—Ä–ł–Ķ–ľ–į –ď–Ľ–ł–≤–Ķ–ļ–į —É –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö —Ā –≤—č—Ā–ĺ–ļ–ł–ľ —Ä–ł—Ā–ļ–ĺ–ľ —Ä–Ķ—Ü–ł–ī–ł–≤–į –≤–Ĺ–ĺ–≤—Ć —Ā—ā–į–Ľ–ł –ĺ—ā–ľ–Ķ—á–į—ā—Ć—Ā—Ź —Ä–Ķ—Ü–ł–ī–ł–≤—č –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź. –ü—Ä–ł —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł–ł —Ā –≥—Ä—É–Ņ–Ņ–ĺ–Ļ –ł—Ā—ā–ĺ—Ä–ł—á–Ķ—Ā–ļ–ĺ–≥–ĺ –ļ–ĺ–Ĺ—ā—Ä–ĺ–Ľ—Ź –į–ī—ä—é–≤–į–Ĺ—ā–Ĺ–į—Ź —ā–Ķ—Ä–į–Ņ–ł—Ź –ī–ĺ—Ā—ā–ĺ–≤–Ķ—Ä–Ĺ–ĺ —É–Ľ—É—á—ą–į–Ľ–į –Ĺ–Ķ —ā–ĺ–Ľ—Ć–ļ–ĺ –Ī–Ķ–∑—Ä–Ķ—Ü–ł–ī–ł–≤–Ĺ—É—é, –Ĺ–ĺ –ł –ĺ–Ī—Č—É—é –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā—Ć —É –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö —Ā –≤—č—Ā–ĺ–ļ–ł–ľ —Ä–ł—Ā–ļ–ĺ–ľ —Ä–Ķ—Ü–ł–ī–ł–≤–į. –ė—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į—ā–Ķ–Ľ–ł –Ņ—Ä–Ķ–ī–Ņ–ĺ–Ľ–ĺ–∂–ł–Ľ–ł, —á—ā–ĺ –Ņ–į—Ü–ł–Ķ–Ĺ—ā–į–ľ —Ā –≤—č—Ā–ĺ–ļ–ł–ľ —Ä–ł—Ā–ļ–ĺ–ľ –Ņ–ĺ–ļ–į–∑–į–Ĺ–į –Ī–ĺ–Ľ–Ķ–Ķ –Ņ—Ä–ĺ–ī–ĺ–Ľ–∂–ł—ā–Ķ–Ľ—Ć–Ĺ–į—Ź —ā–Ķ—Ä–į–Ņ–ł—Ź –ď–Ľ–ł–≤–Ķ–ļ–ĺ–ľ –≤ –į–ī—ä—é–≤–į–Ĺ—ā–Ĺ–ĺ–ľ —Ä–Ķ–∂–ł–ľ–Ķ. –£—á–ł—ā—č–≤–į—Ź –≤—č—Ā–ĺ–ļ—É—é —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć –ď–Ľ–ł–≤–Ķ–ļ–į –≤ —É–Ľ—É—á—ą–Ķ–Ĺ–ł–ł –Ī–Ķ–∑—Ä–Ķ—Ü–ł–ī–ł–≤–Ĺ–ĺ–Ļ –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā–ł, –≤ –ī–Ķ–ļ–į–Ī—Ä–Ķ 2008 –≥. –≤ –°–®–ź –ł –≤ 2009 –≥. –≤ –ē–≤—Ä–ĺ–Ņ–Ķ –ł –†–ĺ—Ā—Ā–ł–ł –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā –ĺ–ī–ĺ–Ī—Ä–Ķ–Ĺ –≤ –ļ–į—á–Ķ—Ā—ā–≤–Ķ –į–ī—ä—é–≤–į–Ĺ—ā–Ĺ–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł –Ņ—Ä–ł CD117-–Ņ–ĺ–Ľ–ĺ–∂–ł—ā–Ķ–Ľ—Ć–Ĺ—č—Ö –≥–į—Ā—ā—Ä–ĺ–ł–Ĺ—ā–Ķ—Ā—ā–ł–Ĺ–į–Ľ—Ć–Ĺ—č—Ö —Ā—ā—Ä–ĺ–ľ–į–Ľ—Ć–Ĺ—č—Ö –ĺ–Ņ—É—Ö–ĺ–Ľ—Ź—Ö. –°—É–Ī–Ņ–ĺ–Ņ—É–Ľ—Ź—Ü–ł–ĺ–Ĺ–Ĺ—č–Ļ –į–Ĺ–į–Ľ–ł–∑, –Ņ—Ä–Ķ–ī—Ā—ā–į–≤–Ľ–Ķ–Ĺ–Ĺ—č–Ļ –Ĺ–į ASCO –≤ 2010 –≥., –Ņ–ĺ–ļ–į–∑–į–Ľ, —á—ā–ĺ –Ĺ–į–ł–Ī–ĺ–Ľ—Ć—ą—É—é –≤—č–≥–ĺ–ī—É –ĺ—ā –į–ī—ä—é–≤–į–Ĺ—ā–Ĺ–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł –≤ –Ņ–Ľ–į–Ĺ–Ķ —É–Ľ—É—á—ą–Ķ–Ĺ–ł—Ź –Ī–Ķ–∑—Ä–Ķ—Ü–ł–ī–ł–≤–Ĺ–ĺ–Ļ –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā–ł –Ņ–ĺ–Ľ—É—á–į—é—ā –Ī–ĺ–Ľ—Ć–Ĺ—č–Ķ —Ā –ľ—É—ā–į—Ü–ł–Ķ–Ļ –≤ 11-–ľ —ć–ļ–∑–ĺ–Ĺ–Ķ [33].
–í –Ķ–≤—Ä–ĺ–Ņ–Ķ–Ļ—Ā–ļ–ĺ–ľ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–ł (EORTC 62024) –ĺ–ļ–ĺ–Ľ–ĺ 900 –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ –Ņ–ĺ—Ā–Ľ–Ķ —Ä–į–ī–ł–ļ–į–Ľ—Ć–Ĺ–ĺ–Ļ –ĺ–Ņ–Ķ—Ä–į—Ü–ł–ł –Ņ–ĺ –Ņ–ĺ–≤–ĺ–ī—É GIST —Ā –≤—č—Ā–ĺ–ļ–ł–ľ –ł –Ņ—Ä–ĺ–ľ–Ķ–∂—É—ā–ĺ—á–Ĺ—č–ľ —Ä–ł—Ā–ļ–ĺ–ľ —Ä–Ķ—Ü–ł–ī–ł–≤–į –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź —Ä–į–Ĺ–ī–ĺ–ľ–ł–∑–ł—Ä–ĺ–≤–į–Ľ–ł –≤ 2 –≥—Ä—É–Ņ–Ņ—č: –≥—Ä—É–Ņ–Ņ–į –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö, –Ņ–ĺ–Ľ—É—á–į–≤—ą–ł—Ö –į–ī—ä—é–≤–į–Ĺ—ā–Ĺ—É—é —ā–Ķ—Ä–į–Ņ–ł—é 400 –ľ–≥ –ď–Ľ–ł–≤–Ķ–ļ–į –≤ —Ā—É—ā–ļ–ł –≤ —ā–Ķ—á–Ķ–Ĺ–ł–Ķ 2 –Ľ–Ķ—ā, —Ā—Ä–į–≤–Ĺ–ł–≤–į–Ľ–į—Ā—Ć —Ā –≥—Ä—É–Ņ–Ņ–ĺ–Ļ –Ĺ–į–Ī–Ľ—é–ī–Ķ–Ĺ–ł—Ź, –Ņ–į—Ü–ł–Ķ–Ĺ—ā—č –ļ–ĺ—ā–ĺ—Ä–ĺ–Ļ –Ņ–ĺ–Ľ—É—á–į–Ľ–ł –Ņ–Ľ–į—Ü–Ķ–Ī–ĺ. –ö—Ä–ĺ–ľ–Ķ —ā–ĺ–≥–ĺ, –Ņ–Ľ–į–Ĺ–ł—Ä–ĺ–≤–į–Ľ–ĺ—Ā—Ć –ł–∑—É—á–ł—ā—Ć –ĺ–Ī—Č—É—é –ł –Ī–Ķ–∑—Ä–Ķ—Ü–ł–ī–ł–≤–Ĺ—É—é –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā—Ć. –Ě–į–Ī–ĺ—Ä –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö –Ĺ–į—á–į–Ľ—Ā—Ź –≤ 2004 –≥. –ł –∑–į–ļ–ĺ–Ĺ—á–ł–Ľ—Ā—Ź –≤ 2008 –≥. –†–Ķ–∑—É–Ľ—Ć—ā–į—ā—č –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź –ĺ–∂–ł–ī–į—é—ā—Ā—Ź –≤ –Ī–Ľ–ł–∂–į–Ļ—ą–Ķ–Ķ –≤—Ä–Ķ–ľ—Ź.
–ü–Ķ—Ä–≤–ł—á–Ĺ–į—Ź –ł –≤—ā–ĺ—Ä–ł—á–Ĺ–į—Ź —Ä–Ķ–∑–ł—Ā—ā–Ķ–Ĺ—ā–Ĺ–ĺ—Ā—ā—Ć –ļ –ď–Ľ–ł–≤–Ķ–ļ—É
–í —ā–Ķ—Ä–į–Ņ–ł–ł GIST —Ä–į–∑–Ľ–ł—á–į—é—ā –Ņ–Ķ—Ä–≤–ł—á–Ĺ—É—é –ł –≤—ā–ĺ—Ä–ł—á–Ĺ—É—é —Ä–Ķ–∑–ł—Ā—ā–Ķ–Ĺ—ā–Ĺ–ĺ—Ā—ā—Ć. –ě—ā—Ā—É—ā—Ā—ā–≤–ł–Ķ —á—É–≤—Ā—ā–≤–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ—Ā—ā–ł –ļ –ď–Ľ–ł–≤–Ķ–ļ—É –≤ —ā–Ķ—á–Ķ–Ĺ–ł–Ķ –Ņ–Ķ—Ä–≤—č—Ö 6 –ľ–Ķ—Ā. —Ā—á–ł—ā–į–Ķ—ā—Ā—Ź –Ņ–Ķ—Ä–≤–ł—á–Ĺ–ĺ–Ļ —Ä–Ķ–∑–ł—Ā—ā–Ķ–Ĺ—ā–Ĺ–ĺ—Ā—ā—Ć—é. –ě–Ĺ–į –ĺ—ā–ľ–Ķ—á–į–Ķ—ā—Ā—Ź –Ņ—Ä–ł–Ī–Ľ–ł–∑–ł—ā–Ķ–Ľ—Ć–Ĺ–ĺ —É 10% –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö. –ü–Ķ—Ä–≤—č–Ķ –∂–Ķ –Ņ—Ä–ĺ—ā–ĺ–ļ–ĺ–Ľ—č –į–Ĺ–į–Ľ–ł–∑–ł—Ä–ĺ–≤–į–Ľ–ł —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć —ā–Ķ—Ä–į–Ņ–ł–ł –≤ –∑–į–≤–ł—Ā–ł–ľ–ĺ—Ā—ā–ł –ĺ—ā –ľ—É—ā–į—Ü–ł–ĺ–Ĺ–Ĺ–ĺ–≥–ĺ —Ā—ā–į—ā—É—Ā–į. –Ď—č–Ľ–ĺ –ĺ—ā–ľ–Ķ—á–Ķ–Ĺ–ĺ, —á—ā–ĺ —ā–ł–Ņ –ľ—É—ā–į—Ü–ł–ł –≤–Ľ–ł—Ź–Ķ—ā –Ĺ–į –Ņ–Ķ—Ä–≤–ł—á–Ĺ—É—é —Ä–Ķ–∑–ł—Ā—ā–Ķ–Ĺ—ā–Ĺ–ĺ—Ā—ā—Ć: –Ņ—Ä–ł –ľ—É—ā–į—Ü–ł–ł –≤ 11-–ľ —ć–ļ–∑–ĺ–Ĺ–Ķ ‚Äď 5%, –≤ 9-–ľ —ć–ļ–∑–ĺ–Ĺ–Ķ ‚Äď 16%, –Ņ—Ä–ł –ī–ł–ļ–ĺ–ľ —ā–ł–Ņ–Ķ ‚Äď 2‚Äď3% [34]. –í —ā–ĺ –∂–Ķ –≤—Ä–Ķ–ľ—Ź –ĺ—ā–Ĺ–ĺ—Ā–ł—ā–Ķ–Ľ—Ć–Ĺ–į—Ź —Ä–Ķ–∑–ł—Ā—ā–Ķ–Ĺ—ā–Ĺ–ĺ—Ā—ā—Ć –ĺ–Ņ—É—Ö–ĺ–Ľ–Ķ–Ļ —Ā –ľ—É—ā–į—Ü–ł–Ķ–Ļ –≤ 9-–ľ —ć–ļ–∑–ĺ–Ĺ–Ķ –ľ–ĺ–∂–Ķ—ā –Ī—č—ā—Ć —Ā–≤—Ź–∑–į–Ĺ–į —Ā –Ĺ–Ķ–ī–ĺ—Ā—ā–į—ā–ĺ—á–Ĺ–ĺ–Ļ –ī–ĺ–∑–ĺ–Ļ –ď–Ľ–ł–≤–Ķ–ļ–į [20]. –ė—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź in vitro –ł –ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ł–Ķ —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā—č –Ņ–ĺ–ļ–į–∑–į–Ľ–ł, —á—ā–ĺ –Ī–ĺ–Ľ—Ć–Ĺ—č–Ķ —Ā –ľ—É—ā–į—Ü–ł–Ķ–Ļ D842V –į–Ī—Ā–ĺ–Ľ—é—ā–Ĺ–ĺ –Ĺ–Ķ —á—É–≤—Ā—ā–≤–ł—ā–Ķ–Ľ—Ć–Ĺ—č –ļ –ď–Ľ–ł–≤–Ķ–ļ—É –ł –≤ —Ā–Ľ—É—á–į–Ķ –ľ–Ķ—ā–į—Ā—ā–į—ā–ł—á–Ķ—Ā–ļ–ĺ–Ļ –Ī–ĺ–Ľ–Ķ–∑–Ĺ–ł –ł–ľ–Ķ—é—ā –Ņ–Ľ–ĺ—Ö–ĺ–Ļ –Ņ—Ä–ĺ–≥–Ĺ–ĺ–∑. –ü–į—Ü–ł–Ķ–Ĺ—ā—č —Ā –ī—Ä—É–≥–ł–ľ–ł –ľ—É—ā–į—Ü–ł—Ź–ľ–ł –≤ –≥–Ķ–Ĺ–Ķ PDGFRA –≤ –Ī–ĺ–Ľ—Ć—ą–ł–Ĺ—Ā—ā–≤–Ķ —Ā–Ľ—É—á–į–Ķ–≤ —á—É–≤—Ā—ā–≤–ł—ā–Ķ–Ľ—Ć–Ĺ—č –ļ —ā–Ķ—Ä–į–Ņ–ł–ł –ł –ī–Ķ–ľ–ĺ–Ĺ—Ā—ā—Ä–ł—Ä—É—é—ā –Ņ—Ä–ĺ–ī–ĺ–Ľ–∂–ł—ā–Ķ–Ľ—Ć–Ĺ—č–Ļ —ć—Ą—Ą–Ķ–ļ—ā –Ĺ–į –ď–Ľ–ł–≤–Ķ–ļ–Ķ. –ü–ĺ—Ā–ļ–ĺ–Ľ—Ć–ļ—É –Ņ—Ä–ł –ī–ł–ļ–ĺ–ľ —ā–ł–Ņ–Ķ –Ĺ–į–Ļ–ī–Ķ–Ĺ—č –ł–Ĺ—č–Ķ –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ—č –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł–ł, —ā–ĺ –Ņ—Ä–Ķ–ī–Ņ–ĺ–Ľ–į–≥–į–Ķ—ā—Ā—Ź, —á—ā–ĺ —ć—Ą—Ą–Ķ–ļ—ā –ľ–ĺ–∂–Ķ—ā –Ĺ–į—Ā—ā—É–Ņ–ł—ā—Ć –ĺ—ā –ī—Ä—É–≥–ł—Ö —ā–į—Ä–≥–Ķ—ā–Ĺ—č—Ö –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤: –ł–Ĺ–≥–ł–Ī–ł—ā–ĺ—Ä–ĺ–≤ VEGFR ‚Äď –ī–Ľ—Ź –Ņ–Ķ–ī–ł–į—ā—Ä–ł—á–Ķ—Ā–ļ–ł—Ö GIST, —Ā—É–ļ—Ü–ł–Ĺ–į—ā–ī–Ķ–≥–ł–ī—Ä–ĺ–≥–Ķ–Ĺ–į–∑—č ‚Äď –ī–Ľ—Ź SDH-–ľ—É—ā–į–Ĺ—ā–Ĺ—č—Ö, BRAF ‚Äď –ī–Ľ—Ź BRAF-–ľ—É—ā–į–Ĺ—ā–Ĺ—č—Ö GIST [35].¬†
–Ě–Ķ—Ā–ľ–ĺ—ā—Ä—Ź –Ĺ–į –≤—č—Ā–ĺ–ļ—É—é —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć —ā–Ķ—Ä–į–Ņ–ł–ł –ď–Ľ–ł–≤–Ķ–ļ–ĺ–ľ –Ņ—Ä–ł –ĺ—ā—Ā—É—ā—Ā—ā–≤–ł–ł –Ņ–Ķ—Ä–≤–ł—á–Ĺ–ĺ–Ļ —Ä–Ķ–∑–ł—Ā—ā–Ķ–Ĺ—ā–Ĺ–ĺ—Ā—ā–ł, —É –Ņ–ĺ–ī–į–≤–Ľ—Ź—é—Č–Ķ–≥–ĺ –Ī–ĺ–Ľ—Ć—ą–ł–Ĺ—Ā—ā–≤–į –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö —Ä–į–∑–≤–ł–≤–į–Ķ—ā—Ā—Ź –≤—ā–ĺ—Ä–ł—á–Ĺ–į—Ź —Ä–Ķ–∑–ł—Ā—ā–Ķ–Ĺ—ā–Ĺ–ĺ—Ā—ā—Ć –ļ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā—É. –í—ā–ĺ—Ä–ł—á–Ĺ—č–Ķ –ľ—É—ā–į—Ü–ł–ł —Ä–į–∑–≤–ł–≤–į—é—ā—Ā—Ź –≤ —ā–ĺ–ľ –∂–Ķ —Ā–į–ľ–ĺ–ľ –≥–Ķ–Ĺ–Ķ, —á—ā–ĺ –ł –Ņ–Ķ—Ä–≤–ł—á–Ĺ—č–Ķ –ľ—É—ā–į—Ü–ł–ł. –ü—Ä–ł –ī–ł–ļ–ĺ–ľ —ā–ł–Ņ–Ķ –≤—ā–ĺ—Ä–ł—á–Ĺ—č–Ķ –ľ—É—ā–į—Ü–ł–ł –Ĺ–Ķ –≤—č—Ź–≤–Ľ—Ź—é—ā—Ā—Ź. –Ď–ĺ–Ľ—Ć—ą–ł–Ĺ—Ā—ā–≤–ĺ –≤—ā–ĺ—Ä–ł—á–Ĺ—č—Ö –ľ—É—ā–į—Ü–ł–Ļ KIT –Ņ—Ä–Ķ–ī—Ā—ā–į–≤–Ľ–Ķ–Ĺ—č —ā–ĺ—á–Ķ—á–Ĺ—č–ľ–ł –∑–į–ľ–Ķ–Ĺ–į–ľ–ł –ĺ–ī–Ĺ–ĺ–≥–ĺ –Ĺ—É–ļ–Ľ–Ķ–ĺ—ā–ł–ī–į, –ļ–ĺ—ā–ĺ—Ä—č–Ķ –Ľ–ĺ–ļ–į–Ľ–ł–∑—É—é—ā—Ā—Ź –≤ —ā–ł—Ä–ĺ–∑–ł–Ĺ–ļ–ł–Ĺ–į–∑–Ĺ—č—Ö –ī–ĺ–ľ–Ķ–Ĺ–į—Ö KIT –ł PDGFRA –ł –≤ –ļ–ł–Ĺ–į–∑-—Ā–≤—Ź–∑—č–≤–į—é—Č–ł—Ö –ī–ĺ–ľ–Ķ–Ĺ–į—Ö KIT (15-–Ļ –ł 16-–Ļ —ć–ļ–∑–ĺ–Ĺ—č). –ö–į–ļ –Ņ—Ä–į–≤–ł–Ľ–ĺ, –≤—ā–ĺ—Ä–ł—á–Ĺ—č–Ķ –ľ—É—ā–į—Ü–ł–ł –ĺ—ā–Ľ–ł—á–į—é—ā—Ā—Ź –ĺ—ā –Ņ–Ķ—Ä–≤–ł—á–Ĺ—č—Ö. –≠—ā–ł –ľ—É—ā–į—Ü–ł–ł –ľ–ĺ–≥—É—ā –ĺ–Ņ—Ä–Ķ–ī–Ķ–Ľ—Ź—ā—Ć—Ā—Ź –ļ–į–ļ –Ķ–ī–ł–Ĺ–ł—á–Ĺ—č–Ķ –ł–Ľ–ł –ļ–į–ļ –Ĺ–Ķ—Ā–ļ–ĺ–Ľ—Ć–ļ–ĺ –ļ–Ľ–ĺ–Ĺ–ĺ–≤ —Ā–ĺ —Ā–Ņ–Ķ—Ü–ł—Ą–ł—á–Ķ—Ā–ļ–ĺ–Ļ –ľ—É—ā–į—Ü–ł–Ķ–Ļ –≤ –ĺ–ī–Ĺ–ĺ–ľ –ĺ–Ņ—É—Ö–ĺ–Ľ–Ķ–≤–ĺ–ľ —É–∑–Ľ–Ķ. –°¬†–ļ–Ľ–ł–Ĺ–ł—á–Ķ—Ā–ļ–ĺ–Ļ —ā–ĺ—á–ļ–ł –∑—Ä–Ķ–Ĺ–ł—Ź –≤–į–∂–Ĺ–ĺ –ĺ—ā–ľ–Ķ—ā–ł—ā—Ć, —á—ā–ĺ –ļ–ĺ–Ľ–ł—á–Ķ—Ā—ā–≤–ĺ –≤—ā–ĺ—Ä–ł—á–Ĺ—č—Ö –ľ—É—ā–į—Ü–ł–Ļ –ľ–ĺ–∂–Ķ—ā –ī–ĺ—Ā—ā–ł–≥–į—ā—Ć 7 —É –ĺ–ī–Ĺ–ĺ–≥–ĺ –Ņ–į—Ü–ł–Ķ–Ĺ—ā–į –≤ —Ä–į–∑–Ľ–ł—á–Ĺ—č—Ö –ľ–Ķ—ā–į—Ā—ā–į–∑–į—Ö [36]. –í—č—Ä–į–∂–Ķ–Ĺ–Ĺ–į—Ź –≥–Ķ—ā–Ķ—Ä–ĺ–≥–Ķ–Ĺ–Ĺ–ĺ—Ā—ā—Ć –ĺ–Ņ—É—Ö–ĺ–Ľ–Ķ–≤—č—Ö –ļ–Ľ–ĺ–Ĺ–ĺ–≤ –∑–į—Ā—ā–į–≤–ł–Ľ–į –ĺ—ā–ļ–į–∑–į—ā—Ć—Ā—Ź –ĺ—ā –Ņ—É–Ĺ–ļ—Ü–ł–ł –ľ–Ķ—ā–į—Ā—ā–į–∑–ĺ–≤ –Ņ—Ä–ł –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł–ł –ĺ–Ņ—É—Ö–ĺ–Ľ–ł –Ĺ–į —Ą–ĺ–Ĺ–Ķ –ď–Ľ–ł–≤–Ķ–ļ–į —Ā —Ü–Ķ–Ľ—Ć—é –ĺ–Ņ—Ä–Ķ–ī–Ķ–Ľ–Ķ–Ĺ–ł—Ź ¬ę–ľ–ł—ą–Ķ–Ĺ–ł¬Ľ –Ņ—Ä–ł –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł–ł. –ü—Ä–ł –ī–ł–ļ–ĺ–ľ —ā–ł–Ņ–Ķ –Ņ—Ä–ł–≤–Ľ–Ķ–ļ–į—ā–Ķ–Ľ—Ć–Ĺ–ĺ–Ļ –ľ–ł—ą–Ķ–Ĺ—Ć—é –ľ–ĺ–∂–Ķ—ā –Ī—č—ā—Ć IGF1R, –≥–ł–Ņ–Ķ—Ä—ć–ļ—Ā–Ņ—Ä–Ķ—Ā—Ā–ł—Ź –ļ–ĺ—ā–ĺ—Ä–ĺ–≥–ĺ –Ĺ–į–Ī–Ľ—é–ī–į–Ķ—ā—Ā—Ź –Ņ—Ä–ł –ī–į–Ĺ–Ĺ–ĺ–ľ –≤–ł–ī–Ķ GIST –ł –Ĺ–Ķ –∑–į–≤–ł—Ā–ł—ā –ĺ—ā KIT-–ľ—É—ā–į—Ü–ł–ł [37].
–ü—Ä–Ķ–Ņ–į—Ä–į—ā—č –≤—ā–ĺ—Ä–ĺ–Ļ –Ľ–ł–Ĺ–ł–ł —ā–Ķ—Ä–į–Ņ–ł–ł –ł –Ĺ–ĺ–≤—č–Ķ –Ņ–ĺ–ī—Ö–ĺ–ī—č
–Ē–Ľ—Ź –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā –Ņ–Ķ—Ä–≤–ł—á–Ĺ–ĺ–Ļ –ł–Ľ–ł –≤—ā–ĺ—Ä–ł—á–Ĺ–ĺ–Ļ —Ä–Ķ–∑–ł—Ā—ā–Ķ–Ĺ—ā–Ĺ–ĺ—Ā—ā—Ć—é –ļ –ď–Ľ–ł–≤–Ķ–ļ—É –Ķ–ī–ł–Ĺ—Ā—ā–≤–Ķ–Ĺ–Ĺ—č–ľ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–ľ –≤—ā–ĺ—Ä–ĺ–Ļ –Ľ–ł–Ĺ–ł–ł –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź —Ā—É–Ĺ–ł—ā–ł–Ĺ–ł–Ī (–°—É—ā–Ķ–Ĺ—ā). –°—É–Ĺ–ł—ā–ł–Ĺ–ł–Ī ‚Äď –Ņ–Ķ—Ä–ĺ—Ä–į–Ľ—Ć–Ĺ—č–Ļ –ł–Ĺ–≥–ł–Ī–ł—ā–ĺ—Ä —ā–ł—Ä–ĺ–∑–ł–Ĺ–ļ–ł–Ĺ–į–∑ —Ā —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć—é –≤ –ĺ—ā–Ĺ–ĺ—ą–Ķ–Ĺ–ł–ł KIT, —Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–į —Ā–ĺ—Ā—É–ī–ł—Ā—ā–ĺ–≥–ĺ —Ą–į–ļ—ā–ĺ—Ä–į —Ä–ĺ—Ā—ā–į (VEGFR), —ā—Ä–ĺ–ľ–Ī–ĺ—Ü–ł—ā–į—Ä–Ĺ–ĺ–≥–ĺ —Ą–į–ļ—ā–ĺ—Ä–į —Ä–ĺ—Ā—ā–į (PDGFR), —Ä–Ķ—Ü–Ķ–Ņ—ā–ĺ—Ä–į –ľ–į–ļ—Ä–ĺ—Ą–į–≥–į–Ľ—Ć–Ĺ–ĺ–≥–ĺ –ļ–ĺ–Ľ–ĺ–Ĺ–ł–Ķ—Ā—ā–ł–ľ—É–Ľ–ł—Ä—É—é—Č–Ķ–≥–ĺ —Ą–į–ļ—ā–ĺ—Ä–į (CSF-1R). –ė—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź —Ä–į–Ĺ–Ĺ–ł—Ö —Ą–į–∑ –Ņ–ĺ –Ņ—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł—é –°—É—ā–Ķ–Ĺ—ā–į –Ņ–ĺ–ļ–į–∑–į–Ľ–ł —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć –≤ –ĺ—ā–Ĺ–ĺ—ą–Ķ–Ĺ–ł–ł GIST —Ā –Ī–ĺ–Ľ—Ć—ą–ł–Ĺ—Ā—ā–≤–ĺ–ľ –≤–ł–ī–ĺ–≤ –ľ—É—ā–į—Ü–ł–Ļ [38]. –ü–ĺ—Ā–Ľ–Ķ–ī–Ĺ–ł–Ķ –ī–į–Ĺ–Ĺ—č–Ķ –Ņ–ĺ–ļ–į–∑–į–Ľ–ł, —á—ā–ĺ –ľ–Ķ–ī–ł–į–Ĺ—č –ī–ĺ –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź –ł –ĺ–Ī—Č–Ķ–Ļ –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā–ł –Ī—č–Ľ–ł –ī–ĺ–Ľ—Ć—ą–Ķ —É –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā –ľ—É—ā–į—Ü–ł–Ķ–Ļ –≤ 9-–ľ —ć–ļ–∑–ĺ–Ĺ–Ķ –ł –ī–ł–ļ–ł–ľ —ā–ł–Ņ–ĺ–ľ –Ņ–ĺ —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—é —Ā –Ņ–į—Ü–ł–Ķ–Ĺ—ā–į–ľ–ł —Ā –ľ—É—ā–į—Ü–ł–Ķ–Ļ –≤ 11-–ľ —ć–ļ–∑–ĺ–Ĺ–Ķ [39]. –í¬†–ļ—Ä—É–Ņ–Ĺ–ĺ–ľ —Ä–į–Ĺ–ī–ĺ–ľ–ł–∑–ł—Ä–ĺ–≤–į–Ĺ–Ĺ–ĺ–ľ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł–ł III —Ą–į–∑—č –ł–∑—É—á–į–Ľ–į—Ā—Ć —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć –°—É—ā–Ķ–Ĺ—ā–į –≤ —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł–ł —Ā –Ņ–Ľ–į—Ü–Ķ–Ī–ĺ —Ā—Ä–Ķ–ī–ł 312 –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā –Ņ–Ķ—Ä–≤–ł—á–Ĺ–ĺ–Ļ —Ä–Ķ–∑–ł—Ā—ā–Ķ–Ĺ—ā–Ĺ–ĺ—Ā—ā—Ć—é –ł–Ľ–ł –Ĺ–Ķ–Ņ–Ķ—Ä–Ķ–Ĺ–ĺ—Ā–ł–ľ–ĺ—Ā—ā—Ć—é –ď–Ľ–ł–≤–Ķ–ļ–į.¬†
–ú–Ķ–ī–ł–į–Ĺ–į –≤—Ä–Ķ–ľ–Ķ–Ĺ–ł –ī–ĺ –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź (TTP) —Ā–ĺ—Ā—ā–į–≤–ł–Ľ–į 27,3 –Ĺ–Ķ–ī–Ķ–Ľ–ł –ī–Ľ—Ź –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤, –Ņ–ĺ–Ľ—É—á–į—é—Č–ł—Ö –°—É—ā–Ķ–Ĺ—ā, –ł 6,4 –Ĺ–Ķ–ī–Ķ–Ľ–ł –ī–Ľ—Ź –≥—Ä—É–Ņ–Ņ—č –Ņ–Ľ–į—Ü–Ķ–Ī–ĺ (p < 0,0001). –í –Ľ–Ķ—á–Ķ–Ī–Ĺ–ĺ–Ļ –≥—Ä—É–Ņ–Ņ–Ķ –Ĺ–į–Ī–Ľ—é–ī–į–Ľ–ĺ—Ā—Ć 7% —á–į—Ā—ā–ł—á–Ĺ—č—Ö –ĺ—ā–≤–Ķ—ā–ĺ–≤, 58% —Ā—ā–į–Ī–ł–Ľ–ł–∑–į—Ü–ł–Ļ –ł –ī–ĺ—Ā—ā–ĺ–≤–Ķ—Ä–Ĺ–ĺ —á–į—Č–Ķ –ĺ–Ī—ä–Ķ–ļ—ā–ł–≤–Ĺ—č–Ļ –ĺ—ā–≤–Ķ—ā –Ņ–ĺ —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—é —Ā –≥—Ä—É–Ņ–Ņ–ĺ–Ļ –Ņ–Ľ–į—Ü–Ķ–Ī–ĺ. –†–Ķ–∑—É–Ľ—Ć—ā–į—ā—č –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź –Ņ–ĺ—Ā–Ľ—É–∂–ł–Ľ–ł –ĺ—Ā–Ĺ–ĺ–≤–ĺ–Ļ –ī–Ľ—Ź –ĺ–ī–ĺ–Ī—Ä–Ķ–Ĺ–ł—Ź –°—É—ā–Ķ–Ĺ—ā–į –≤ –ļ–į—á–Ķ—Ā—ā–≤–Ķ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į –≤—ā–ĺ—Ä–ĺ–Ļ –Ľ–ł–Ĺ–ł–ł –Ņ—Ä–ł –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł–ł –Ņ–ĺ—Ā–Ľ–Ķ –ď–Ľ–ł–≤–Ķ–ļ–į –ł–Ľ–ł –Ņ–Ķ—Ä–≤–ł—á–Ĺ–ĺ–Ļ –Ĺ–Ķ–Ņ–Ķ—Ä–Ķ–Ĺ–ĺ—Ā–ł–ľ–ĺ—Ā—ā–ł –ł–ľ–į—ā–ł–Ĺ–ł–Ī–į [40]. –ě–ī–Ĺ–ł–ľ –ł–∑ —Ā—É—Č–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ—č—Ö –Ĺ–Ķ–≥–į—ā–ł–≤–Ĺ—č—Ö –ĺ—ā–Ľ–ł—á–ł–Ļ –°—É—ā–Ķ–Ĺ—ā–į –ĺ—ā –ď–Ľ–ł–≤–Ķ–ļ–į —Ź–≤–ł–Ľ–į—Ā—Ć –Ī–ĺ–Ľ–Ķ–Ķ –≤—č—Ä–į–∂–Ķ–Ĺ–Ĺ–į—Ź —ā–ĺ–ļ—Ā–ł—á–Ĺ–ĺ—Ā—ā—Ć, –Ņ—Ä–ĺ—Ź–≤–Ľ—Ź—é—Č–į—Ź—Ā—Ź –Ľ–Ķ–Ļ–ļ–ĺ–Ņ–Ķ–Ĺ–ł–Ķ–Ļ –ł —ā—Ä–ĺ–ľ–Ī–ĺ—Ü–ł—ā–ĺ–Ņ–Ķ–Ĺ–ł–Ķ–Ļ —É –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤. –≠—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć –°—É—ā–Ķ–Ĺ—ā–į –ī–Ľ—Ź –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö —Ā –ľ—É—ā–į—Ü–ł–Ķ–Ļ –≤ 13-–ľ –ł 14-–ľ —ć–ļ–∑–ĺ–Ĺ–į—Ö –Ī—č–Ľ–į –≤—č—ą–Ķ, —á–Ķ–ľ —Ā –ľ—É—ā–į—Ü–ł–Ķ–Ļ –≤ 17-–ľ —ć–ļ–∑–ĺ–Ĺ–Ķ [39].
P. Rutkowski –ł —Ā–ĺ–į–≤—ā. –≤ –Ĺ–Ķ–ī–į–≤–Ĺ–Ķ–ľ —Ā–ĺ–ĺ–Ī—Č–Ķ–Ĺ–ł–ł –Ņ—Ä–Ķ–ī—Ā—ā–į–≤–ł–Ľ–ł –ī–į–Ĺ–Ĺ—č–Ķ –ĺ–ī–Ĺ–ĺ—Ü–Ķ–Ĺ—ā—Ä–ĺ–≤–ĺ–≥–ĺ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź, –≤–ļ–Ľ—é—á–į–≤—ą–Ķ–≥–ĺ 137 –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤ —Ā –ľ–Ķ—ā–į—Ā—ā–į—ā–ł—á–Ķ—Ā–ļ–ĺ–Ļ —Ą–ĺ—Ä–ľ–ĺ–Ļ GIST –°—É—ā–Ķ–Ĺ—ā–ĺ–ľ –Ņ–ĺ—Ā–Ľ–Ķ –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź –Ĺ–į —Ą–ĺ–Ĺ–Ķ —ā–Ķ—Ä–į–Ņ–ł–ł –ď–Ľ–ł–≤–Ķ–ļ–ĺ–ľ [41]. –ě–ī–Ĺ–ĺ–≥–ĺ–ī–ł—á–Ĺ–į—Ź –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā—Ć –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö —Ā–ĺ—Ā—ā–į–≤–ł–Ľ–į 42%. –ě–Ī—Č–į—Ź –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā—Ć, —Ä–į—Ā—Ā—á–ł—ā–į–Ĺ–Ĺ–į—Ź –ĺ—ā –Ĺ–į—á–į–Ľ–į –Ņ—Ä–ł–Ķ–ľ–į –ď–Ľ–ł–≤–Ķ–ļ–į –ł –°—É—ā–Ķ–Ĺ—ā–į, —Ā–ĺ—Ā—ā–į–≤–ł–Ľ–į —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ 74 –Ĺ–Ķ–ī–Ķ–Ľ–ł –ł 51 –ľ–Ķ—Ā—Ź—Ü. –õ—É—á—ą–ł–Ļ –ĺ—ā–≤–Ķ—ā –Ĺ–į —ā–Ķ—Ä–į–Ņ–ł—é –°—É—ā–Ķ–Ĺ—ā–ĺ–ľ –ĺ—ā–ľ–Ķ—á–Ķ–Ĺ —É –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö —Ā –ľ—É—ā–į—Ü–ł–Ķ–Ļ –≤ 9-–ľ —ć–ļ–∑–ĺ–Ĺ–Ķ –ł –Ņ—Ä–ł –ī–ł–ļ–ĺ–ľ —ā–ł–Ņ–Ķ (–ľ–Ķ–ī–ł–į–Ĺ–į –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā–ł 65,5 –ł 50,5 –Ĺ–Ķ–ī–Ķ–Ľ–ł) –Ņ–ĺ —Ā—Ä–į–≤–Ĺ–Ķ–Ĺ–ł—é —Ā –ľ—É—ā–į—Ü–ł–Ķ–Ļ –≤ 11-–ľ —ć–ļ–∑–ĺ–Ĺ–Ķ –ł–Ľ–ł PDGFRA (36,8 –ł 9 –Ĺ–Ķ–ī–Ķ–Ľ—Ć —Ā–ĺ–ĺ—ā–≤–Ķ—ā—Ā—ā–≤–Ķ–Ĺ–Ĺ–ĺ). –ö—Ä–ĺ–ľ–Ķ —ā–ĺ–≥–ĺ, –≤—ā–ĺ—Ä—č–ľ –Ĺ–Ķ–∑–į–≤–ł—Ā–ł–ľ—č–ľ —Ą–į–ļ—ā–ĺ—Ä–ĺ–ľ –Ņ—Ä–ĺ–≥–Ĺ–ĺ–∑–į, –ī–ĺ—Ā—ā–ĺ–≤–Ķ—Ä–Ĺ–ĺ –≤–Ľ–ł—Ź—é—Č–ł–ľ –Ĺ–į –Ī–Ķ–∑—Ä–Ķ—Ü–ł–ī–ł–≤–Ĺ—É—é –ł –ĺ–Ī—Č—É—é –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā—Ć –≤ –ĺ–ī–Ĺ–ĺ- –ł –ľ–Ĺ–ĺ–≥–ĺ—Ą–į–ļ—ā–ĺ—Ä–Ĺ–ĺ–ľ –į–Ĺ–į–Ľ–ł–∑–Ķ, —Ź–≤–ł–Ľ–į—Ā—Ć –į—Ä—ā–Ķ—Ä–ł–į–Ľ—Ć–Ĺ–į—Ź –≥–ł–Ņ–Ķ—Ä—ā–Ķ–Ĺ–∑–ł—Ź: –Ņ–į—Ü–ł–Ķ–Ĺ—ā—č, —É –ļ–ĺ—ā–ĺ—Ä—č—Ö —Ą–ł–ļ—Ā–ł—Ä–ĺ–≤–į–Ľ—Ā—Ź –ī–į–Ĺ–Ĺ—č–Ļ –Ņ–ĺ–Ī–ĺ—á–Ĺ—č–Ļ —ć—Ą—Ą–Ķ–ļ—ā —ā–Ķ—Ä–į–Ņ–ł–ł –°—É—ā–Ķ–Ĺ—ā–ĺ–ľ, –∂–ł–Ľ–ł –ī–ĺ—Ā—ā–ĺ–≤–Ķ—Ä–Ĺ–ĺ –ī–ĺ–Ľ—Ć—ą–Ķ.
–Ē—Ä—É–≥–ł–Ķ —ā–į—Ä–≥–Ķ—ā–Ĺ—č–Ķ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā—č, –ł–Ĺ–≥–ł–Ī–ł—Ä—É—é—Č–ł–Ķ —ā–ł—Ä–ĺ–∑–ł–Ĺ–ļ–ł–Ĺ–į–∑—č (–Ĺ–ĺ –Ĺ–Ķ VEGFR), ‚Äď –ľ–į–∑–į—ā–ł–Ĺ–ł–Ī –ł –ī–į–∑–į—ā–ł–Ĺ–ł–Ī. –ė–Ĺ–≥–ł–Ī–ł—ā–ĺ—Ä—č —ā–ł—Ä–ĺ–∑–ł–Ĺ–ļ–ł–Ĺ–į–∑, –į–ļ—ā–ł–≤–Ĺ—č–Ķ –Ĺ–Ķ —ā–ĺ–Ľ—Ć–ļ–ĺ –≤ –ĺ—ā–Ĺ–ĺ—ą–Ķ–Ĺ–ł–ł KIT –ł PDGFR, –Ĺ–ĺ –ł –Ņ–ĺ–ī–į–≤–Ľ—Ź—é—Č–ł–Ķ VEGFR: —Ā–ĺ—Ä–į—Ą–Ķ–Ĺ–ł–Ī, –ľ–ĺ—ā–Ķ–∑–į–Ĺ–ł–Ī, –≤–į—ā–į–Ľ–į–Ĺ–ł–Ī ‚Äď –Ņ—Ä–ĺ—Ö–ĺ–ī—Ź—ā –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź –≤—ā–ĺ—Ä–ĺ–Ļ —Ą–į–∑—č. –Ď–Ķ–≤–į—Ü–ł–∑—É–ľ–į–Ī ‚Äď –ľ–ĺ–Ĺ–ĺ–ļ–Ľ–ĺ–Ĺ–į–Ľ—Ć–Ĺ–ĺ–Ķ –į–Ĺ—ā–ł—ā–Ķ–Ľ–ĺ, –Ī–Ľ–ĺ–ļ–ł—Ä—É—é—Č–Ķ–Ķ VEGFR, ‚Äď –ł–∑—É—á–į–Ķ—ā—Ā—Ź –≤ –ļ–į—á–Ķ—Ā—ā–≤–Ķ –ī–ĺ–Ņ–ĺ–Ľ–Ĺ–Ķ–Ĺ–ł—Ź –ļ –ď–Ľ–ł–≤–Ķ–ļ—É –≤ III —Ą–į–∑–Ķ –ł—Ā—Ā–Ľ–Ķ–ī–ĺ–≤–į–Ĺ–ł—Ź. –ė–∑—É—á–į—é—ā—Ā—Ź –ł –Ľ–Ķ–ļ–į—Ä—Ā—ā–≤–į —Ā –Ĺ–Ķ–Ņ—Ä—Ź–ľ—č–ľ –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ–ĺ–ľ –ī–Ķ–Ļ—Ā—ā–≤–ł—Ź ‚Äď –ł–Ĺ–≥–ł–Ī–ł—ā–ĺ—Ä—č –Ī–Ķ–Ľ–ļ–į —ā–Ķ–Ņ–Ľ–ĺ–≤–ĺ–≥–ĺ —ą–ĺ–ļ–į (IPI-504) –ł –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ–į PI3K/AKT/mTOR (—ć–≤–Ķ—Ä–ĺ–Ľ–ł–ľ—É—Ā). –ú–Ĺ–ĺ–≥–ĺ–ĺ–Ī—Ä–į–∑–ł–Ķ –≤—č—ą–Ķ—É–ļ–į–∑–į–Ĺ–Ĺ—č—Ö —ā–į—Ä–≥–Ķ—ā–Ĺ—č—Ö –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–≤ –≥–ĺ–≤–ĺ—Ä–ł—ā –ĺ —Ā–Ľ–Ķ–ī—É—é—Č–Ķ–ľ. –Ě–Ķ—Ā–ľ–ĺ—ā—Ä—Ź –Ĺ–į —ć—Ą—Ą–Ķ–ļ—ā–ł–≤–Ĺ–ĺ—Ā—ā—Ć –ĺ–ī–Ĺ–ĺ–≥–ĺ —ā–į—Ä–≥–Ķ—ā–Ĺ–ĺ–≥–ĺ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į (–ď–Ľ–ł–≤–Ķ–ļ), —Ä–į–Ĺ–ĺ –ł–Ľ–ł –Ņ–ĺ–∑–ī–Ĺ–ĺ –ĺ–Ņ—É—Ö–ĺ–Ľ—Ć –Ĺ–į—Ö–ĺ–ī–ł—ā –Ĺ–ĺ–≤—č–Ķ –Ņ—É—ā–ł —Ä–į–∑–≤–ł—ā–ł—Ź. –í–ĺ–∑–Ĺ–ł–ļ–į–Ķ—ā¬† –Ĺ–Ķ–ĺ–Ī—Ö–ĺ–ī–ł–ľ–ĺ—Ā—ā—Ć –Ī–Ľ–ĺ–ļ–ł—Ä–ĺ–≤–į–Ĺ–ł—Ź —ć—ā–ł—Ö –Ņ—É—ā–Ķ–Ļ –ī—Ä—É–≥–ł–ľ–ł —ā–į—Ä–≥–Ķ—ā–Ĺ—č–ľ–ł –į–≥–Ķ–Ĺ—ā–į–ľ–ł (—ā–į–Ī–Ľ. 2). –ü–ĺ—Ā–ļ–ĺ–Ľ—Ć–ļ—É –Ņ–ĺ—ā–Ķ–Ĺ—Ü–ł–į–Ľ—Ć–Ĺ—č—Ö –Ņ—É—ā–Ķ–Ļ ¬ę—É—Ā–ļ–ĺ–Ľ—Ć–∑–į–Ĺ–ł—Ź¬Ľ –∑–į–Ī–ĺ–Ľ–Ķ–≤–į–Ĺ–ł—Ź –ī–ĺ—Ā—ā–į—ā–ĺ—á–Ĺ–ĺ –ľ–Ĺ–ĺ–≥–ĺ, —Ā–Ņ–ł—Ā–ĺ–ļ –≤–ĺ–∑–ľ–ĺ–∂–Ĺ—č—Ö –Ľ–Ķ–ļ–į—Ä—Ā—ā–≤ —ā–į–ļ–∂–Ķ —Ä–į—Ā—ą–ł—Ä—Ź–Ķ—ā—Ā—Ź, –Ľ–ł–Ī–ĺ –Ĺ—É–∂–Ĺ–ĺ –ĺ—ā–ī–į–≤–į—ā—Ć –Ņ—Ä–Ķ–ī–Ņ–ĺ—á—ā–Ķ–Ĺ–ł–Ķ –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā—É —Ā –ľ–Ĺ–ĺ–∂–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ—č–ľ –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ–ĺ–ľ –ī–Ķ–Ļ—Ā—ā–≤–ł—Ź, –Ĺ–ĺ —ā–ĺ–≥–ī–į –Ņ–ĺ–Ĺ—Ź—ā–ł–Ķ ¬ę—ā–į—Ä–≥–Ķ—ā–Ĺ–į—Ź —ā–Ķ—Ä–į–Ņ–ł—Ź¬Ľ –≤ –ł–∑–≤–Ķ—Ā—ā–Ĺ–ĺ–Ļ –ľ–Ķ—Ä–Ķ —Ä–į–∑–ľ—č–≤–į–Ķ—ā—Ā—Ź. –ö—Ä–ĺ–ľ–Ķ —ā–ĺ–≥–ĺ, –ľ–Ĺ–ĺ–∂–Ķ—Ā—ā–≤–Ķ–Ĺ–Ĺ—č–Ļ –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ –ī–Ķ–Ļ—Ā—ā–≤–ł—Ź –Ĺ–Ķ–ł–∑–Ī–Ķ–∂–Ĺ–ĺ –≤–Ķ–ī–Ķ—ā –∑–į —Ā–ĺ–Ī–ĺ–Ļ –Ī–ĺ–Ľ–Ķ–Ķ —ā—Ź–∂–Ķ–Ľ—č–Ķ –Ņ–ĺ–Ī–ĺ—á–Ĺ—č–Ķ —ć—Ą—Ą–Ķ–ļ—ā—č –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–į.
–ó–į–ļ–Ľ—é—á–Ķ–Ĺ–ł–Ķ
–Ē–Ķ—Ā—Ź—ā–ł–Ľ–Ķ—ā–Ĺ–ł–Ļ –ĺ–Ņ—č—ā –Ņ—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł—Ź —ā–į—Ä–≥–Ķ—ā–Ĺ–ĺ–Ļ —ā–Ķ—Ä–į–Ņ–ł–ł –≥–į—Ā—ā—Ä–ĺ–ł–Ĺ—ā–Ķ—Ā—ā–ł–Ĺ–į–Ľ—Ć–Ĺ—č—Ö —Ā—ā—Ä–ĺ–ľ–į–Ľ—Ć–Ĺ—č—Ö –ĺ–Ņ—É—Ö–ĺ–Ľ–Ķ–Ļ –Ņ—Ä–ĺ–ī–Ķ–ľ–ĺ–Ĺ—Ā—ā—Ä–ł—Ä–ĺ–≤–į–Ľ –Ĺ–Ķ–Ī—č–≤–į–Ľ—č–Ļ —É—Ā–Ņ–Ķ—Ö –≤ –ĺ–Ĺ–ļ–ĺ–Ľ–ĺ–≥–ł–ł. –í–Ņ–Ķ—Ä–≤—č–Ķ –≤ –ī–ł–į–≥–Ĺ–ĺ—Ā—ā–ł–ļ–Ķ –ł –Ľ–Ķ—á–Ķ–Ĺ–ł–ł —Ā–ĺ–Ľ–ł–ī–Ĺ—č—Ö –ĺ–Ņ—É—Ö–ĺ–Ľ–Ķ–Ļ —É–ī–į–Ľ–ĺ—Ā—Ć –Ĺ–Ķ —ā–ĺ–Ľ—Ć–ļ–ĺ –≤—č—Ź–≤–ł—ā—Ć –ļ–Ľ—é—á–Ķ–≤–ĺ–Ļ –ľ–Ķ—Ö–į–Ĺ–ł–∑–ľ –Ņ–į—ā–ĺ–≥–Ķ–Ĺ–Ķ–∑–į, –Ĺ–ĺ –ł –Ņ–ĺ–ī–ĺ–Ī—Ä–į—ā—Ć –≤—č—Ā–ĺ–ļ–ĺ—ā–ĺ—á–Ĺ–ĺ–Ķ ¬ę–ĺ—Ä—É–∂–ł–Ķ¬Ľ –ļ ¬ę–ľ–ł—ą–Ķ–Ĺ–ł¬Ľ (–į–Ĺ–≥–Ľ. target ‚Äď –ľ–ł—ą–Ķ–Ĺ—Ć). –ü—Ä–ł–ľ–Ķ–Ĺ–Ķ–Ĺ–ł–Ķ –ď–Ľ–ł–≤–Ķ–ļ–į —É–Ľ—É—á—ą–ł–Ľ–ĺ –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā—Ć –Ī–ĺ–Ľ—Ć–Ĺ—č—Ö —Ā –ľ–Ķ—ā–į—Ā—ā–į—ā–ł—á–Ķ—Ā–ļ–ĺ–Ļ —Ą–ĺ—Ä–ľ–ĺ–Ļ –Ī–ĺ–Ľ–Ķ–∑–Ĺ–ł –≤ —ā—Ä–ł —Ä–į–∑–į. –ü—Ä–Ķ–ī–ĺ–Ņ–Ķ—Ä–į—Ü–ł–ĺ–Ĺ–Ĺ–į—Ź —ā–Ķ—Ä–į–Ņ–ł—Ź –Ņ–ĺ–∑–≤–ĺ–Ľ–ł–Ľ–į —É–Ľ—É—á—ą–ł—ā—Ć —Ä–Ķ–∑—É–Ľ—Ć—ā–į—ā—č –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź –Ņ—Ä–ł –ľ–Ķ—Ā—ā–Ĺ–ĺ—Ä–į—Ā–Ņ—Ä–ĺ—Ā—ā—Ä–į–Ĺ–Ķ–Ĺ–Ĺ—č—Ö —Ą–ĺ—Ä–ľ–į—Ö. –ź–ī—ä—é–≤–į–Ĺ—ā–Ĺ–į—Ź —ā–Ķ—Ä–į–Ņ–ł—Ź —Ā–Ĺ–ł–∑–ł–Ľ–į —Ä–ł—Ā–ļ —Ä–Ķ—Ü–ł–ī–ł–≤–į –ł —É–≤–Ķ–Ľ–ł—á–ł–Ľ–į –ĺ–Ī—Č—É—é –≤—č–∂–ł–≤–į–Ķ–ľ–ĺ—Ā—ā—Ć –Ņ–į—Ü–ł–Ķ–Ĺ—ā–ĺ–≤. –°—ā—Ä–ĺ–ľ–į–Ľ—Ć–Ĺ—č–Ķ –ĺ–Ņ—É—Ö–ĺ–Ľ–ł –ĺ–Ī–Ľ–į–ī–į—é—ā –≤—č—Ä–į–∂–Ķ–Ĺ–Ĺ–ĺ–Ļ –≥–Ķ—ā–Ķ—Ä–ĺ–≥–Ķ–Ĺ–Ĺ–ĺ—Ā—ā—Ć—é –Ņ–ĺ –ľ—É—ā–į—Ü–ł–ĺ–Ĺ–Ĺ–ĺ–ľ—É —Ā—ā–į—ā—É—Ā—É, –ļ–ĺ—ā–ĺ—Ä—č–Ļ —Ź–≤–Ľ—Ź–Ķ—ā—Ā—Ź —Ą–į–ļ—ā–ĺ—Ä–ĺ–ľ –Ņ—Ä–ĺ–≥–Ĺ–ĺ–∑–į –ł, –Ņ–ĺ –≤–ĺ–∑–ľ–ĺ–∂–Ĺ–ĺ—Ā—ā–ł, –ī–ĺ–Ľ–∂–Ķ–Ĺ –≤—č–Ņ–ĺ–Ľ–Ĺ—Ź—ā—Ć—Ā—Ź –≤—Ā–Ķ–ľ –Ī–ĺ–Ľ—Ć–Ĺ—č–ľ. –†–į–∑–≤–ł—ā–ł–Ķ –≤—ā–ĺ—Ä–ł—á–Ĺ—č—Ö –ľ—É—ā–į—Ü–ł–Ļ –ł –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł—Ä–ĺ–≤–į–Ĺ–ł–Ķ –Ĺ–į —Ą–ĺ–Ĺ–Ķ –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź –Ņ—Ä–Ķ–Ņ–į—Ä–į—ā–ĺ–ľ –Ņ–Ķ—Ä–≤–ĺ–Ļ –Ľ–ł–Ĺ–ł–ł –ī–ł–ļ—ā—É—é—ā –Ĺ–Ķ–ĺ–Ī—Ö–ĺ–ī–ł–ľ–ĺ—Ā—ā—Ć –ł–∑–ľ–Ķ–Ĺ–Ķ–Ĺ–ł—Ź —ā–į–ļ—ā–ł–ļ–ł –Ľ–Ķ—á–Ķ–Ĺ–ł—Ź –ł –Ņ–ĺ–ł—Ā–ļ–į –Ĺ–ĺ–≤—č—Ö –Ņ—É—ā–Ķ–Ļ –Ī–Ľ–ĺ–ļ–į–ī—č –ĺ–Ņ—É—Ö–ĺ–Ľ–Ķ–≤–ĺ–Ļ –Ņ—Ä–ĺ–≥—Ä–Ķ—Ā—Ā–ł–ł.