Клиническое наблюдение наследственного панкреатита у ребенка в результате мутации в гене PRSS1
- Аннотация
- Статья
- Ссылки
- English
Введение
Хронический панкреатит (ХП) – хроническое заболевание поджелудочной железы (ПЖ) различной этиологии, протекающее с повторными эпизодами паренхиматозного воспаления, приводящими к необратимым морфологическим изменениям паренхимы и ее замещению фиброзной тканью. Фибротическая реорганизация ткани поджелудочной железы способствует прогрессирующему снижению ее экзокринной и эндокринной функции и может сопровождаться формированием кист, псевдокист, стриктур, конкрементов, обструкцией протоковой системы, сосудистыми осложнениями, болевым синдромом, мальнутрицией. Как следствие – снижение качества и продолжительности жизни [1].
К основным этиологическим причинам формирования ХП у взрослых относят употребление алкоголя и желчнокаменную болезнь (ЖКБ) [1–4]. При алкогольном панкреатите повреждение ПЖ обусловлено гиперсекрецией белка ацинарными клетками. В протоковой системе белковый секрет, не сбалансированный гиперпродукцией воды и бикарбонатов, накапливается в мелких протоках в виде белковых преципитатов. Впоследствии формируются конкременты ПЖ с отложениями солей кальция. Подобные изменения приводят к развитию интра- и перидуктального склероза, локальных стенозов и обтурации протоков ПЖ с одновременной дилатацией протоковой системы. Еще один фактор, участвующий в патогенезе алкогольного панкреатита, – изменение тонуса сфинктера Одди. Спазм сфинктера Одди вызывает внутрипротоковую гипертензию, а релаксация способствует рефлюксу дуоденального содержимого и внутрипротоковой активации панкреатических ферментов [2].
Курение – независимый фактор прогрессирования ХП [4–6]. Хронический панкреатит у курящих наблюдается в два раза чаще, чем у некурящих. Риск развития заболевания зависит от количества выкуриваемых сигарет [4, 7, 8]. Курение истощает запасы витаминов C и А, а также снижает сывороточный уровень других антиоксидантов, что приводит к повреждению ткани железы свободными радикалами [9].
Нередко ХП становится результатом дуоденопанкреатического рефлюкса, возникающего на фоне дисфункции сфинктера Одди и ЖКБ, особенно при наличии дуоденальной гипертензии. Гипертензия в протоке ПЖ, связанная с обструкцией протока или ампулы дуоденального сосочка, впоследствии приводит к разрыву мелких панкреатических протоков, выделению секрета в паренхиму железы и активации пищеварительных ферментов. Обычно рецидивы билиарного панкреатита возникают при миграции конкрементов размером до 4 мм. Наличие в желчном пузыре конкрементов менее 5 мм в диаметре увеличивает риск развития панкреатита в четыре раза [10].
Высокий риск панкреатита наблюдается при повышении уровня триглицеридов более 500 мг/дл. Механизм развития ХП обусловлен токсическим воздействием на ткань ПЖ высоких концентраций свободных жирных кислот, которые не могут быть полностью связаны сывороточными альбуминами в плазме крови [2].
Хронический панкреатит у 12% пациентов с первичным гиперпаратиреозом обусловлен травматическим повреждением железы из-за гиперкальциемии [11, 12].
Сахарный диабет 1-го и 2-го типов служит независимым фактором риска развития панкреатита. В свою очередь сахарный диабет 3-го типа (панкреатогенный) может быть следствием ХП. Связь между ХП и сахарным диабетом 3-го типа обусловлена деструкцией островков Лангерганса на фоне прогрессирующих воспалительных изменений в ткани ПЖ. Такие изменения влекут за собой снижение продукции инсулина, соматостатина, глюкагона, а следовательно, развитие диабета [13].
Причинами развития ХП могут быть вирусная инфекция (гепатит B, вирусы гриппа, Коксаки и др.), длительный прием лекарственных средств (глюкокортикостероидов, нестероидных противовоспалительных препаратов, цитостатиков, эстрогенов), нарушение кровообращения и травмы ПЖ.
Аутоиммунная этиология панкреатита – редкая форма хронических заболеваний ПЖ. Это системное воспалительное заболевание, при котором ПЖ является одним из органов-мишеней. Впервые аутоиммунный панкреатит был описан в 1961 г. [14].
В крупном исследовании K. Yoshida и соавт. [15] с участием 731 пациента у мужчин аутоиммунный панкреатит встречался чаще, чем у женщин (2:1). У пациентов с подозрением на карциному ПЖ, получивших оперативное лечение, в 50% случаев гистологически подтвердился диагноз аутоиммунного панкреатита [16]. При обследовании пациентов выявлен повышенный уровень иммуноглобулина (Ig) G (за счет фракции IgG4), у незначительного количества пациентов обнаружены антитела к лактоферрину и карбоангидразе. Такие антитела при аутоиммунном панкреатите выявляются редко, поэтому они не относятся к актуальным диагностическим критериям. В свою очередь повышение уровня IgG4-антител отмечалось примерно у 50% пациентов с подозрением на панкреатит аутоиммунной этиологии. При гистологическом исследовании аутоиммунный панкреатит характеризуется плотной перидуктальной лимфоплазмоцитарной инфильтрацией с явлениями облитерирующего флебита и перидуктальным фиброзом (1-й тип аутоиммунного панкреатита) или гранулоцитарным повреждением эпителия (GELs) в ПЖ и схожим повреждением других органов (2-й тип аутоиммунного панкреатита) [17]. Аутоиммунный панкреатит 2-го типа манифестирует преимущественно у женщин, ассоциирован с воспалительными заболеваниями кишечника и редко имеет рецидивирующее течение. Для этого типа аутоиммунного панкреатита не характерно повышение IgG4-антител в сыворотке крови [17].
Диагностика аутоиммунного панкреатита основана на критериях HiSORT. Речь идет о гистологических, серологических критериях, аутоиммунном поражении других органов и систем и ответе на стероидную терапию [18, 19].
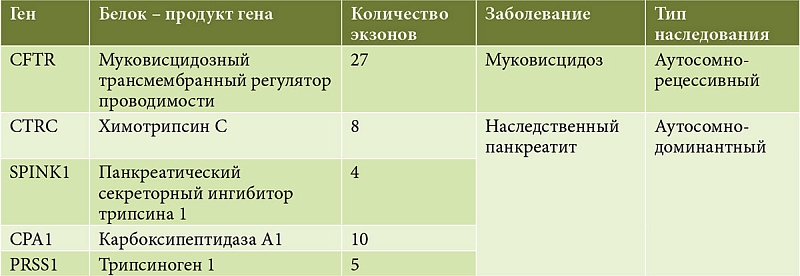
В настоящее время все больше внимания уделяется изучению генетических факторов – триггеров хронического воспаления ПЖ. К основным генам, мутации в которых приводят к формированию ХП, можно отнести CFTR, PRSS1, SPINK1, CTRC и CPA1 [20–25]. Общая характеристика протестированных генов представлена в таблице [26]. Мутации в указанных генах существенно увеличивают риск развития ХП.
Ген PRSS1 обеспечивает синтез трипсиногена 1, на долю которого в составе общего трипсиногена ПЖ приходится 2/3. Мутации в гене PRSS1 приводят к развитию ХП с аутосомно-доминантным типом наследования [27, 28]. Ген PRSS1 обладает неполной пенетрантностью – около 80%. Это означает, что примерно у 20% людей, несущих патогенную мутацию PRSS1, заболевание клинически не проявляется, вероятно из-за наличия дополнительных генетических факторов, в том числе имеющих протективное значение, в других генах, влияющих на работу ПЖ. К наиболее частым и повторяющимся мутациям гена PRSS1 относятся Arg122His (c.365G>A), Arg122Cys (c.364C>T), Asn29Ile (c.86A>T), Asn29Thr (c.86A>C), Arg116Cys (c.346C>T), Ala16Val (c.47C>T) и Glu79Lys (c.235G>A). Трипсиноген является ключевой молекулой в патогенезе панкреатита. До 66% пациентов с наследственным панкреатитом имеют мутацию в гене PRSS1. Распространенность наследственного панкреатита составляет в среднем 0,3:100 000 [27–29].
Следует отметить, что для развития панкреатита недостаточно генетической предрасположенности. Необходим инициирующий внешний фактор (злоупотребление алкоголем, наличие билиарной патологии и др.), способствующий манифестации заболевания.
Ген SPINK1 кодирует ингибитор трипсина. Физиологическая роль данного белка сводится к предотвращению катализируемого трипсином процесса активации зимогенов в ПЖ. Мутации в гене SPINK1 у пациентов с идиопатическим и наследственным панкреатитом приводят к увеличению активности трипсина в ПЖ. Так запускается патологический воспалительный процесс в паренхиме органа. Для SPINK1-ассоциированного панкреатита характерен аутосомно-доминантный тип наследования.
К частым мутациям гена SPINK1 относят Asn34Ser (до 13% больных), Leu14Arg и c.194+2T>C. Первая из представленных мутаций (Asn34Ser) достаточно распространена в мире, особенно в южноазиатских популяциях, где ее частота достигает 2%. В целом мутации в гене SPINK1 встречаются у 30% пациентов с идиопатическим хроническим панкреатитом и только у 1–2% общей популяции [30, 31].
У 25–30% пациентов с идиопатическим ХП выявляется гетерозиготное носительство мутации в гене CFTR. Ген CFTR кодирует муковисцидозный трансмембранный регулятор проводимости и обеспечивает работу хлорного канала слизеобразующих желез, посредством которого происходит транспорт ионов натрия и хлора через клеточную мембрану. В классическом варианте (при наличии мутаций CFTR в гомозиготной или компаунд-гетерозиготной форме) патогенные изменения данного гена приводят к развитию наследственного аутосомно-рецессивного заболевания муковисцидоза. Однако гетерозиготное носительство мутаций CFTR считается генетическим фактором риска развития гипоферментного панкреатита [23].
Другим генетическим фактором развития ХП служат изменения гена СTRC. Первые данные о мутациях в CTRC появились в 2008 г. [32]. Сегодня известно, что ген CTRC кодирует химотрипсин C, физиологическая роль которого заключается в интрапанкреатической деградации трипсина и трипсиногена. Мутации в гене CTRC нарушают блокировку чрезмерной интрапанкреатической активации трипсина с помощью химотрипсина С. Мутации в гене CTRC выявляются у 3,3% пациентов с идиопатическим панкреатитом [32, 33].
В 2013 г. была установлена еще одна генетическая причина панкреатита – патогенные варианты гена CPA1. Ген кодирует выработку фермента карбоксипептидазы А1. Показано, что функционально значимые варианты гена CPA1 в гетерозиготной форме существенно увеличивают риск развития ХП. Относительный риск его развития в группе пациентов с манифестацией до десяти лет особенно высок – 84. Считается, что мутации в гене CPA1 ведут к нарушению функции эндоплазматического ретикулума в клетках ПЖ и запрограммированной гибели ацинарных клеток [34].
Клинический случай
Пациент 9 лет с непрерывно-рецидивирующим ХП, у которого при проведении молекулярно-генетического исследования выявлена мутация в гене PRSS1.
Анамнез жизни. Ребенок от второй физиологически протекавшей беременности, вторых естественных срочных родов. Масса при рождении – 5300 г, длина – 51 см. Грудное вскармливание до шести месяцев, затем – искусственное. Раннее развитие без особенностей, у специалистов на диспансерном учете не состоял.
Анамнез заболевания. Впервые заболел в возрасте 5 лет: жалобы на рвоту, диарею до шести раз в сутки, повышение температуры до субфебрильных цифр. Из эпидемиологического анамнеза известно, что ребенок выезжал в Южный федеральный округ, употреблял в пищу сырое фермерское молоко. При обследовании инфекционистом исключены вирусные гепатиты А, B, C, бактериальные кишечные инфекции (сальмонеллез, шигеллез, эшерихиоз). На фоне симптоматического лечения (антациды, ферменты) отмечалась положительная динамика, однако через месяц вновь возник эпизод повышения температуры, сопровождавшийся рвотой, болями в животе, диарейным синдромом. Ребенок обследован по месту жительства.
Ультразвуковое исследование (УЗИ) брюшной полости: гепатоспленомегалия, лимфоаденопатия, свободная жидкость в брюшной полости с взвесью. В лабораторных исследованиях – изолированное повышение уровня панкреатической амилазы до трех-четырех норм, уровни аланинаминотрансферазы (АЛТ), аспартатаминотрансферазы (АСТ), щелочной фосфатазы (ЩФ), гамма-глутамилтранспептидазы (ГГТП) – в пределах референсных значений. Общий анализ крови – без патологических изменений. Методом иммуноферментного анализа (ИФА) исключены описторхоз, токсокароз, трихинеллез, лямблиоз, амебиаз. В связи с лимфоаденопатией для проведения дифференциальной диагностики между инфекционным и лимфопролиферативным процессом ребенка направили в Московский областной научно-исследовательский клинический институт (МОНИКИ) им. М.Ф. Владимирского. При поступлении обращало на себя внимание вздутие живота, гепатомегалия +2 см по среднеключичной линии, спленомегалия +3,0 см из-под края реберной дуги. Данные лабораторных исследований показали анемию легкой степени (106 г/л), тромбоцитоз (512 тыс.), гипопротеинемию (общий белок 60 г/л), гипоальбуминемию (40 г/л при норме 45–55 г/л), умеренную гипогаммаглобулинемию IgG – 6,2 г/л (норма 7–12 г/л), при нормальных значениях общих IgM, IgA. Биохимические показатели крови: АЛТ, АСТ, ЩФ, ГГТП в пределах референсных значений, за исключением панкреатической амилазы, уровень которой в 1,5–2 раза превышал нормальные значения. В копрологии выявлено большое количество нейтрального жира. Впервые отмечались изменения в коагулограмме крови в виде гипофибриногенемии 0,79–1,67 г/л (норма 2–4 г/л), умеренного повышения уровня антитромбина III 139% (норма 83–128%), тромбинового времени 30 с (15–25 с). Подозрения гематолога на наследственную форму гипофибриногенемии впоследствии не подтвердились.
С помощью визуализирующих методов диагностики (мультиспиральная компьютерная томография) выявлены гепатоспленомегалия, большой лимфоузел в воротах селезенки, лимфоаденопатия в области селезенки, множественные лимфоузлы в брыжейке тонкой кишки. Структура ПЖ не изменена. Умеренный асцит. Лимфоаденопатии в грудной полости не выявлено, в обеих аксиллярных областях множественные лимфоузлы до 10 мм в диаметре. Результаты рекомендованного инфекционистом обследования на туляремию, кампилобактериоз отрицательные. Консультация онколога: данных о лимфопролиферативном процессе нет. Ребенок получал месалазин, нифуроксазид, омепразол, ферментную терапию с умеренным положительным эффектом. Мальчик выписан с диагнозом хронического панкреатита, полиаденопатии.
После выписки из стационара в течение месяца у ребенка сохранялись боли в животе. Через два месяца ребенок повторно поступил в МОНИКИ им. М.Ф. Владимирского. Результаты компьютерной томографии брюшной полости: ПЖ увеличена – 22 ×13 × 12 мм, неровные нечеткие контуры, накопление контрастного препарата снижено преимущественно в головке и теле ПЖ, парапанкреатическая клетчатка уплотнена, инфильтрирована, лимфоузлы увеличены. Вирсунгов проток – 1,5–2,0 мм, стенка двенадцатиперстной кишки и прилежащих отделов тощей кишки утолщена. Лимфоаденопатия по ходу брыжейки тонкой кишки, спленомегалия. Данных об уровне амилазы и липазы крови не представлено. Мальчик обследован на оппортунистические инфекции методом ИФА: герпес 1-го, 2-го, 6-го типов, цитомегаловирус – результат отрицательный, повышены антитела класса IgG к вирусу Эпштейна – Барр до 264 Ед/л (норма до 16 Ед/л). У инфекциониста ребенок не наблюдался, лечения не получал.
В течение двух лет периодически беспокоили умеренные боли в животе, которые купировались спазмолитическими препаратами, курсами получал антациды и панкреатин в минимикросферах.
В сентябре 2016 г. (в возрасте 7 лет) в связи с жалобами на периодические боли в животе и неоформленный стул впервые исследована панкреатическая эластаза кала. Ее уровень составил 168 мкг/г (норма выше 200 мкг/г).
Впоследствии ребенок с жалобами на рвоту и рецидивирующие боли в животе неоднократно госпитализировался в гастроэнтерологическое отделение Московского государственного медицинского университета им. И.М. Сеченова. При клиническом осмотре признаки трофологической недостаточности и гепатоспленомегалия не выявлены. Результаты лабораторного обследования: уровни общего белка, АЛТ, АСТ, общего и прямого билирубина, С-реактивного белка, амилазы, диастазы мочи в пределах референсных значений. Коагулограмма без патологических изменений. Фиброэзофагогастродуоденоскопия показала эрозии желудка, в связи с чем применена трехкомпонентная схема антихеликобактерной терапии.
В возрасте 8 лет с жалобами на приступообразные боли в животе, явлениями асцита ребенок поступил в отделение торакальной хирургии Детской городской клинической больницы № 13 им. Н.Ф. Филатова. При обследовании отмечалось повышение уровня панкреатической амилазы до восьми норм. Результаты исследования крови на наличие вирусов (цитомегаловирус, вирус Эпштейна – Барр, герпес 6-го типа) методом полимеразной цепной реакции отрицательные, на наличие инфекции (токсоплазмоз, эхинококкоз, аскаридоз, лямблии) методом ИФА – также отрицательные. При дополнительном обследовании на панель глистной инвазии и патогенную кишечную группу в Институте медицинской паразитологии, тропических и трансмиссивных заболеваний им. Е.И. Марциновского патологии не выявлено. Гормоны тиреоидного спектра – Т3, Т4, ТТГ – в пределах референсных значений. Показатели гуморального иммунитета (IgG, IgМ, IgА) в пределах нормы. Для исключения IgG4-ассоциированного панкреатита исследован уровень IgG4. Результат в пределах референсных значений. Исследование онкомаркера рака поджелудочной железы СА19-9 – результат отрицательный. Магнитно-резонансная томография выявила признаки хронического панкреатита: неоднородная гиперэхогенная структура ПЖ, с участками отека, множественными мелкими уплотнениями, умеренная спленомегалия, асцит.
После стабилизации состояния, разрешения асцита, расширения энтеральной нагрузки, снижения уровня амилазы с восьми до двух норм ребенок выписан домой. Однако спустя три дня вновь госпитализирован в хирургическое отделение с болевым абдоминальным синдромом. В биохимическом анализе крови – увеличение уровня амилазы до четырех норм. УЗИ органов брюшной полости вновь показало значительное количество свободной жидкости в брюшной полости, изменения структуры ПЖ: увеличение размеров ПЖ 25 × 10 × 26 мм, структура неоднородная, гиперэхогенная с участками отека, умеренное расширение вирсунгова протока до 3,4 мм, спленомегалия 106 × 46 мм.
На консилиуме врачей под руководством члена-корреспондента РАМН, профессора А.Ю. Разумовского принято решение о проведении оперативного лечения ребенку с рецидивирующим течением тяжелого ХП, резистентного к стандартной терапии.
В июне 2017 г. в возрасте 8 лет мальчику провели лапаротомию с наложением панкреатоеюноанастомоза по Ру. Интраоперационно выявлены геморрагический выпот в брюшной полости (300 мл), расширение общего панкреатического протока (до 3 мм). При гистологическом исследовании ткани ПЖ установлен диагноз: хронический панкреатит, массивный фиброз ПЖ, липоматоз, гиперплазия островков Лангерганса. В послеоперационном периоде ребенок получал ингибиторы протеазы, синтетический аналог соматостатина в течение десяти дней, с десятых послеоперационных суток – энтеральное кормление смесью на основе гидролизованного белка молочной сыворотки с постепенным увеличением объема питания и расширением продуктов, согласно диете № 5 по Певзнеру. При неоднократном послеоперационном контроле уровня амилазы данный показатель не превышал референсных значений, в том числе после отмены ингибиторов протеазы и синтетического аналога соматостатина, а также на фоне расширения диеты. В удовлетворительном состоянии на 23-и послеоперационные сутки с уровнем амилазы 49 Ед/л (норма 2280 Ед/л) и липазы 68 Ед/мл (норма 4–130 Ед/мл) ребенок выписан домой.
В течение года после выписки у ребенка дважды отмечались выраженные боли в животе на фоне нарушения диеты. Лечение проходил в амбулаторных условиях.
При УЗИ в августе 2018 г. (в возрасте 9 лет) клинических признаков трофологической недостаточности не выявлено: поджелудочная железа с четкими, неровными контурами, не увеличена в размерах – 16,7 × 7,4 × 15,6 мм, паренхима повышенной эхогенности, неоднородная, без гипоэхогенных участков. Вирсунгов проток в области хвоста не расширен. Копрология: нейтральный жир, жирные кислоты, мыла в умеренном количестве. Панкреатическая эластаза кала 171 мкг/г (норма выше 200 мкг/г). Мать ребенка самостоятельно уменьшила дозу панкреатина. Препарат ребенок получал нерегулярно.
В октябре 2018 г. на фоне интеркуррентного заболевания у мальчика вновь отмечалось обострение панкреатита, потребовавшее госпитализации, интенсивной терапии в течение недели. С учетом рецидивирующего характера панкреатита ребенку рекомендовано генетическое исследование для исключения наследственных форм панкреатита.
Проведено молекулярно-генетическое исследование на наличие мутаций в генах CFTR, CTRC, SPINK1, CPA1 и PRSS1. Метод исследования – массовое параллельное секвенирование (Next Generation Sequencing, NGS) всей колирующей последовательности генов. Для проведения анализа создана праймерная таргетная панель из 65 пар праймеров. Используемая платформа – Ion S5 (Thermo Fisher Scientific). Суммарная область покрытия составила около 13 000 нуклеотидов. Молекулярно-генетическая часть работы проведена на базе Центрального НИИ эпидемиологии Роспотребнадзора.
В ходе исследования у пациента выявлена патогенная мутация c.86A>T (p.Asn29Ile) в гене PRSS1 в гетерозиготной форме.
К настоящему моменту в гене PRSS1 описано свыше 25 различных мутаций. Большинство из них в гетерозиготной форме вызывает развитие аутосомно-доминантной формы панкреатита [26].
Семейный анамнез в таких случаях обычно отягощен панкреатитом. По законам Менделя, теоретическая вероятность передачи заболевания из поколения в поколение 50%. Однако ген PRSS1 обладает неполной пенетрантностью. Показано, что патогенные мутации в гене трипсиногена приводят к проявлению заболевания в 80% случаев. В 20% симптомы отсутствуют (пока по неизвестным причинам) [35–37].
К сожалению, в силу семейных обстоятельств получить достаточную информацию о состоянии здоровья родственников пробанда по отцовской линии не удалось. По материнской линии выявлен единичный случай ХП у матери пациента, у которой ранее, в возрасте 23 лет, диагностирована ЖКБ, проведено оперативное вмешательство в виде холецистэктомии. В настоящее время (возраст 41 год) при УЗИ органов брюшной полости обнаружены диффузные изменения ПЖ (атак острого панкреатита не наблюдалось).
Обсуждение
Обнаруженная у пациента мутация c.86A>T (p.Asn29Ile) является одной из частых мутаций гена PRSS1 [38]. Эта мутация, по данным различных авторов, обнаруживается у 5–21% больных с наследственной формой ХП [36, 39, 40]. В целом патогенное действие мутаций гена PRSS1 обусловлено двумя основными механизмами – преждевременной внутрипанкреатической активацией трипсина или его устойчивостью к деградации. Показано, что in vitro патогенный вариант p.Asn29Ile, выявленный у пациента, сопровождается значительным увеличением аутоактивации катионного трипсиногена [41]. В свою очередь преждевременная активация панкреатических ферментов, еще до выхода сока из протока ПЖ, вызывает воспалительную реакцию и провоцирует процесс аутолиза в отношении собственных ацинарных клеток органа. Впоследствии атаки панкреатита повторяются, и воспалительный процесс приобретает хронический характер.
Сегодня известно, что патогенные мутации гена PRSS1 сопровождаются значительным увеличением риска развития рака ПЖ. В частности, риск развития рака ПЖ у пациентов с наследственным панкреатитом возрастает в 50–70 раз. Риск развития рака ПЖ повышается начиная с 40 лет и достигает 40–70% в возрасте 70 лет (особенно если наследственный панкреатит прослеживается по мужской линии) [20, 42].
С учетом изложенного всем пациентам помимо разнообразных профилактических мер в отношении предупреждения повторных атак панкреатита (избегание стрессовых воздействий, ограничение пищи с высоким содержанием жиров, исключение курения и алкоголя) должен быть рекомендован комплекс мер, направленных на максимально раннее выявление опухоли (мониторинг уровня онкомаркера СА19-9 в крови, регулярное проведение УЗИ поджелудочной железы, магнитно-резонансной томографии и др.).
Заключение
При непрерывно-рецидивирующем течении идиопатического панкреатита после исключения алкогольного, билиарнозависимого, дисметаболического, инфекционного, лекарственного и аутоиммунного генеза заболевания необходимо проведение генетических тестов для выявления наследственных форм панкреатита.
В представленном клиническом наблюдении наследственный панкреатит манифестировал у мальчика в возрасте 5 лет и носил непрерывно-рецидивирующий характер. Результаты проведенного молекулярно-генетического анализа подтвердили наследственный характер ХП (PRSS1-ассоциированный панкреатит). Рекомендовано генетическое тестирование родственников первой и второй степени родства с целью своевременного выявления лиц, предрасположенных к развитию панкреатита.
Все пациенты с генетически подтвержденным наследственным панкреатитом нуждаются в динамическом наблюдении гастроэнтеролога для профилактики и лечения обострений, коррекции экзокринной недостаточности ПЖ, дефицита макро- и микронутриентов, мониторинга раннего выявления онкологического процесса в ПЖ.
Авторы статьи подтвердили отсутствие конфликта интересов и финансовой поддержки исследования, о которой необходимо сообщить.
Ye.A. Gordeyeva, M.G. Ipatova, PhD, A.Yu. Razumovsky, DM, D.V. Zalihin, PhD, Ye.I. Komina, A.D. Matsvay, K.F. Khafizov, M.M. Litvinova, PhD
N.F. Filatov Children’s City Clinical Hospital № 13
Pirogov Russian National Research Medical University
Central Research Institute of Epidemiology of The Federal Service on Customers' Rights Protection and Human Well-Being Surveillance
I.M. Sechenov First Moscow State Medical University
The Loginov Moscow Clinical Scientific Center
Contact person: Yekaterina Aleksandrovna Gordeyeva, ekgordeeva32@gmail.com
Many etiological factors are involved in the development of chronic pancreatitis. In contrast to the adult population, in which this disease development is being more often associated with alcohol and smoking, in Pediatrics, special attention is being paid to genetic causes of disease. Mutations in the genes CFTR, CTRC, SPINK1, CPA1 and PRSS1 significantly increase the risk of this disease developing. The article discusses the case of continuous recurrent chronic pancreatitis in a boy of nine years with a mutation in the PRSS1 gene, revealed during the molecular genetic study.
Уважаемый посетитель uMEDp!
Уведомляем Вас о том, что здесь содержится информация, предназначенная исключительно для специалистов здравоохранения.
Если Вы не являетесь специалистом здравоохранения, администрация не несет ответственности за возможные отрицательные последствия, возникшие в результате самостоятельного использования Вами информации с портала без предварительной консультации с врачом.
Нажимая на кнопку «Войти», Вы подтверждаете, что являетесь врачом или студентом медицинского вуза.