Терапевтическая модуляция микроглии: от молекулярных механизмов к клиническому применению в неврологии
- Аннотация
- Статья
- Ссылки
- English

Введение
Современная неврология переживает этап переосмысления фундаментальных представлений о механизмах нейровоспаления, что обусловлено ограниченной эффективностью терапевтических подходов к лечению пациентов с болезнью Альцгеймера (БА), болезнью Паркинсона (БП), рассеянным склерозом, постинсультными нарушениями, хронической болью. Именно к микроглии как ключевому регулятору нейроимунных процессов в центральной нервной системе (ЦНС) приковано внимание исследователей. Углубленное изучение этих механизмов создает предпосылки для разработки терапевтических стратегий, направленных на селективную нейроиммуномодуляцию микроглиальных функций, что, согласно современным экспериментальным исследованиям, обладает значительным потенциалом. Правда, в настоящее время клинические данные ограничены в количестве и воспроизводимости доклинических результатов.
Цель данной публикации – комплексная систематизация современных подходов к терапевтической модуляции микроглии на основе анализа ее биологии, механизмов активации и болезнь-специфичных нейроиммунных каскадов.
Биология микроглии: основы и современные представления
В 1919 г. исследователь Пио дель Рио-Хортега впервые обнаружил новый тип глиальных клеток. Из-за малого размера сомы он назвал их микроглией. Ученый установил, что клетки микроглии способны выполнять фагоцитарную функцию в отношении дендритных шипиков и клеточного детрита, а также взаимодействовать с другими клетками мозгового вещества. Последние 20 лет исследования микроглии демонстрируют экспоненциальный рост.
Микроглия – резидентные мононуклеарные фагоциты ЦНС, регулирующие развитие мозга, поддержание нейронных сетей и восстановление после повреждений. Микроглия динамична, гетерогенна, может выполнять как нейропротекторные, так и нейротоксичные функции, строго модулируется со стороны микроокружения, поддается перепрограммированию, «стареет», изменяется с возрастом и реагирует на ось «кишечник – микробиота – мозг» [1].
Классически выделяли «покоящуюся» микроглию (М0) и два полярных фенотипа – М1 (провоспалительный, нейротоксичный) и М2 (нейропротекторный). М1-микроглия продуцирует интерлейкин (ИЛ) 1 бета, ИЛ-6, ИЛ-12, фактор некроза опухоли (ФНО) альфа, CCL2 (CC-chemokine ligand 2), активирует пути митоген-активируемой протеинкиназы (MAPK) и ядерного фактора каппа B (NF-kB) и экспрессирует MHC класса II (класс молекул главного комплекса гистосовместимости), Fc-рецепторы, интегрины и костимулирующие молекулы. М2-микроглия выделяет противовоспалительные цитокины (ИЛ-4, ИЛ-13), GDNF, BDNF и поддерживает репаративные процессы. Современные данные демонстрируют существование широкого спектра промежуточных и болезнь-специфичных фенотипов, выходящих за рамки бинарной модели [2].
Анализ транскриптомов (RNA-seq) при БА, амиотрофическом латеральном склерозе и старении позволил выделить болезньассоциированную микроглию (DAM) – подтип, локализующийся вокруг бляшек бета-амилоида и участвующий в их клиренсе [1, 3]. DAM характеризуется сохранением базовых микроглиальных маркеров (iba1, hexb, cst3), снижением экспрессии «гомеостатических» генов (p2ry12, cx3cr1, tmem119, cd33, p2ry13) и повышением экспрессии лизосомальных и липидметаболических генов, включая ctsd, trem2, tyrobp, липопротеинлипазу (Lpl), аполипопротеин E (ApoE). При ишемическом инсульте описан иной профиль микроглии – SAM (stroke-associated microglia), характеризующийся усилением антиоксидантных путей prdx1 – srxn1 – txn1 и специфическим ответом на окислительный стресс [4]. Известно несколько состояний микроглии при БА. Речь, в частности, идет о гомеостатической микроглии, амилоидчувствительной, дистрофической и подвижной [5].
Несмотря на огромное разнообразие микроглии, для всех ее фенотипов характерны определенные феномены. Речь прежде всего идет о постоянном «патрулировании» мозга. Длительная активация вследствие смены преобладающего фенотипа приводит к нейровоспалению, провоспалительные фенотипы и субтипы микроглии отличаются сокращением ветвления. Субпопуляции микроглии формируют не кластеры, а транскрипционный континуум, необходимый для быстрой адаптации к изменениям.
Общие молекулярные механизмы и сигнальные пути активации микроглии
Микроглия человека часто находится в состоянии промежуточной активации (CD68+, сниженная P2RY12), что отличает ее от гомеостатического фенотипа у лабораторных животных [6]. Активация микроглии опосредуется рецепторами врожденного иммунитета и цитокиновыми, пуринергическими и хемокиновыми путями.
Рецептор TREM2, экспрессируемый микроглией, нейронами и астроцитами, распознает фосфатидилсерин, миелиновые обломки, бета-амилоид и ApoE. Его активация индуцирует переход к фаготипу с усилением фагоцитоза, выживания и противовоспалительного ответа, а также участвует в формировании DAM-фенотипа [5, 7].
Рецепторы P2X4, P2X7 и P2Y1 реагируют на внеклеточный аденозинтрифосфат (АТФ). При активации P2X7 кратковременная стимуляция обеспечивает приток Ca2+ и запуск вторичных мессенджеров, а длительная – формирование поры в мембране, запуск инфламмасомы NLRP3, активацию каспазы 1 и созревание ИЛ-1-бета [8].
Рецептор фракталкина CX3CR1, ограниченно экспрессируемый микроглией, регулирует взаимодействие с нейронами и иммунными клетками. Сигналинг по оси CX3CL1 – CX3CR1 способен ослаблять NF-kB-зависимую экспрессию ИЛ-23 и CCL20 и тем самым ограничивать рекрутирование Th17-клеток [9, 10].
Комплементный рецептор 3 (CR3) распознает C3b/iC3b-опсонизированные структуры и участвует в фагоцитозе синапсов и агрегатов. Рецептор к конечным продуктам гликирования (RAGE), связывающий бета-амилоид и другие молекулярные паттерны, ассоциированные с повреждениями (DAMP), активирует NF-kB и усиливает продукцию провоспалительных медиаторов [9].
Толл-подобные рецепторы (TLR1–TLR9) экспрессируются в нейронах, микроглии и астроцитах и участвуют в распознавании сигналов патоген-ассоциированных/дистресс-ассоциированных молекулярных паттернов (PAMP/DAMP). Их активация ассоциирована с БА, БП, боковым амиотрофическим склерозом и инсультом и приводит к запуску NF-kB, MAPK и продукции цитокинов [6].
ИЛ-1-бета через ИЛ-1R активирует NF-kB, усиливая синтез оксид азота (NO), простагландинов и провоспалительных цитокинов [11]. ФНО-альфа, действуя через рецептор R1 ФНО-альфа, запускает NF-kB и в условиях сильного стимула – каспаза-8-зависимый апоптоз [12]. Микроглия опосредует комплемент-зависимый синаптический «прунинг» через C3–CR3, что при патологической активации приводит к избыточной потере синапсов [13]. Понимание этих универсальных путей прайминга микроглии лежит в основе разработки стратегий ее терапевтической модуляции.
Патологически детерминированные сигнальные пути и механизмы, отличающие каждое заболевание
Болезнь-специфичный характер участия микроглии в патогенезе заболеваний ЦНС и уникальный молекулярный профиль при каждом из них открывают возможность создания методов таргетного терапевтического воздействия.
Рассеянный склероз
Рассеянный склероз (РС) – хроническое аутоиммунное заболевание ЦНС, характеризующееся нейровоспалением и нейродегенерацией. Все больше исследований подтверждают важную роль микроглии в развитии данного заболевания.
В активных очагах головного мозга практически все микроглиальные клетки находятся в состоянии активации – снижается экспрессия рецептора P2RY12, TMEM119. Генетические исследования показывают, что гены предрасположенности к РС преимущественно связаны с функциями микроглии, а не нейронов или астроцитов.
Запуск активации микроглии зависит от взаимодействия миелоидных клеток с CD4+ Т-клетками. Последние начинают продуцировать гранулоцитарно-макрофагальный колониестимулирующий фактор (GM-CSF), который активирует микроглию и в сочетании с интерфероном (ИФН) гамма стимулирует дифференцировку моноцитов в воспалительные макрофаги, усиливая повреждение ткани [3].
Дополнительным механизмом усиления иммунопатологии при РС является нарушение баланса между регуляторными Т-клетками (Treg) и провоспалительными Th17-клетками. Показано, что у пациентов с РС Treg обладают повышенной экспрессией TLR2. Активация TLR2 приводит к индукции ИЛ-6, ИФН-гамма, ИЛ-17 и GM-CSF [3].
Активация нейровоспаления начинается с повреждения ткани и привлечения нейтрофилов. Ключевую роль здесь играет путь CXCR2. Через него нейтрофилы могут усиливать повреждение и взаимодействовать с микроглией, что делает CXCR2 потенциальной терапевтической мишенью [14]. В ответ микроглия продуцирует нейротрофические и противовоспалительные факторы, обеспечивая нейропротекцию, однако при длительной активации начинает продуцировать нейротоксичные медиаторы и провоспалительные цитокины, усиливая окислительный стресс.
Кроме того, микроглия регулирует экспрессию BMP-4, ингибирующего ремиелинизацию, а также его антагониста ноггина (noggin), уровень которого повышается в областях успешного восстановления миелина. Таким образом, дисфункция микроглии не только усиливает повреждение, но и препятствует ремиелинизации, способствуя прогрессированию патологического процесса [6].
Болезнь Паркинсона
БП является вторым по распространенности нейродегенеративным заболеванием после БА. Многие гены, связанные с БП, экспрессируются в глиальных клетках и нейронах. Это позволяет предположить, что продукты мутировавших генов в микроглии или нейронах являются частью этиологии заболевания.
Патологические агрегаты альфа-синуклеина (олигомеры, фибриллы) действуют как DAMP-молекулы и активируют микроглию через TLR2, инициируя воспалительный ответ, что является отличительной чертой БП. В микроглии активируется NF-kB-сигнальный путь, индуцируя выраженный провоспалительный фенотип. Альфа-синуклеин связывается с FcγRIIB на поверхности микроглии и снижает ее фагоцитарную активность, что ухудшает клиренс патогенных форм белка. TLR4 также инициирует воспалительный ответ на альфа-синуклеин и участвует в фагоцитарной активности микроглии, способствуя распространению патологического альфа-синуклеина [1]. Активация TLR9 в микроглии вызывает гибель дофаминергических нейронов через глюкокортикоид-зависимый путь [15].
Существуют митохондриально-воспалительные взаимодействия при мутациях генов Parkin и PINK1. Эти мутации усиливают образование активных форм кислорода (ROS), что делает микроглию склонной к гипервоспалительному состоянию. Дефектная митофагия при отсутствии белка Parkin повышает активацию NLRP3-инфламмасомы, усиливая ИЛ-1-бета-зависимую нейротоксичность [16].
Помимо провоспалительной роли микроглия выполняет функцию очистки альфа-синуклеина. Например, CR4 (комплементный рецептор 4) связывается с фибриллярным альфа-синуклеином, индуцируя формирование фаголизосомы и способствуя его удалению [17].
Позитронно-эмиссионная томография (ПЭТ) пациентов с БП выявила широко распространенную активацию микроглии. Примечательно, что она обнаруживается у пациентов не только с длительным течением заболевания, но и с недавно диагностированной патологией [18].
Болезнь Альцгеймера
БА – прогрессирующее нейродегенеративное заболевание, характеризующееся нейропатологическими признаками, такими как внеклеточные бета-амилоидные бляшки и внутриклеточные агрегаты гиперфосфорилированного тау-белка. Активация врожденного иммунитета, синаптическая дисфункция, прогрессирующая гибель нейронов и элиминация перинейрональных сетей в совокупности – характерные проявления деятельности активированной микроглии. Исследования аутопсийных образцов мозга пациентов с БА показали выраженную активацию микроглии при заболевании. Более того, ПЭТ позволяет обнаружить активацию микроглии у пациентов с легкими когнитивными нарушениями. И этот показатель положительно коррелирует с амилоидной нагрузкой [19].
По сравнению с общими воспалительными заболеваниями генетические факторы оказывают более значимое влияние на предрасположенность к БА. Они определяют как количество микроглиальных клеток, так и экспрессию иммуновоспалительных генов в клетках ЦНС.
Обратимся к уникальным клеточным и молекулярным событиям, определяющим активацию микроглии при БА. Так, роль микроглии зависит от стадии заболевания. На ранних этапах она способствует удалению бета-амилоида, тормозит гиперфосфорилирование тау-белка и выделяет нейротрофические факторы, выполняя нейропротективную функцию. По мере прогрессирования заболевания хроническая стимуляция переводит микроглию в устойчивое провоспалительное состояние, что вносит вклад в нейродегенерацию.
Ключевым патогенетическим признаком БА является активация микроглии через широкий спектр рецепторов распознавания молекулярных паттернов: TLR, NLR, RAGE, scavenger-, формилпептидные, комплементные и Fc-рецепторы. Эти рецепторы идентифицируют бета-амилоид и патологический тау-белок как DAMP-сигналы и инициируют внутриклеточные каскады NF-kB, JAK-STAT и NLRP3-инфламмасомы. Взаимодействие TREM2 и ApoE определяет транскрипционный профиль дисфункциональной микроглии и направляет ее по патологической траектории развития [1].
Упоминавшаяся болезнь-ассоциированная микроглия демонстрирует повышение экспрессии генов, связанных с лизосомальными, фагоцитарными и липидными метаболическими путями, включая известные факторы риска развития БА: катепсин D (CtsD), TREM2, Tyrobp, Lpl и ApoE. После распознавания депозитов бета-амилоида микроглия увеличивает экспрессию ИЛ-3Rα, тогда как астроциты выделяют ИЛ-3. Их взаимодействие усиливает миграцию микроглии к амилоидным скоплениям и повышает эффективность удаления бета-амилоида [3].
С возрастом, а также при БА снижается экспрессия рецепторов микроглии, участвующих в распознавании и удалении патологических структур, и формируется фенотип микроглии, характеризующийся сниженной способностью к клиренсу бета-амилоида.
Эффект фагоцитоза бета-амилоида остается неоднозначным: наряду с многочисленными данными, подтверждающими его защитную роль, результаты ряда исследований демонстрируют, что фагоцитоз бета-амилоида микроглией может способствовать формированию амилоидных бляшек [1].
Инсульт
Ишемический инсульт – одна из главных причин тяжелой инвалидности и смертности в мире. Постинсультные нейровоспаление и когнитивные нарушения привлекают все больше внимания в аспекте разработки новых эффективных стратегий лечения.
Микроглия влияет на развитие инсульта с ранних этапов: сначала способствует нейровоспалению, а на поздних стадиях участвует в восстановлении после ишемии благодаря индукции образования глиального рубца.
Дегенерирующие нейроны выделяют сигналы опасности, такие как ламинин, матриксные металлопротеиназы, альфа-синуклеин и нейромеланин. Поэтому считается, что амебоидная микроглия в зоне ишемического ядра поляризована в фенотип M1. Активированная нейротоксичная микроглия высвобождает PGE2, ФНО-альфа, ИЛ-1-бета, NO, ROS и перекись водорода. Эти медиаторы формируют длительные воспалительные каскады, что приводит к усилению повреждения мозга [20].
Как показали экспериментальные исследования инсульта, активация пути Notch индуцирует поляризацию микроглии в фенотип M1, подавляет M2-ответ и усугубляет ишемическое повреждение [21].
Реактивная микроглия может усиливать распространение и токсичность тау-белка через активацию NLRP3-инфламмасомы и сигнального пути NF-kB. Активация инфламмасомы приводит к превращению прокаспазы 1 в активную каспазу 1, которая расщепляет про-ИЛ-1-бета и про-ИЛ-18 до их активных провоспалительных форм и способствует их секреции. Повышение активности каспазы 1 может запускать гибель клеток напрямую через пироптоз или косвенно через апоптоз [21].
Стресс-индуцированная гипералгезия
Рост распространенности хронической боли, не поддающейся традиционному лечению, требует изучения нейроиммунных механизмов патогенеза данного состояния. Исследования подтверждают участие микроглии в формировании гипералгезии, нейропатической и функциональной боли.
Высокий уровень стресса способствует длительной центральной сенситизации. В дорсальном роге спинного мозга повторяющиеся болевые сигналы усиливают высвобождение нейромедиаторов, переводящих микроглию в провоспалительное (M1) состояние. Это вызывает низкоуровневое нейровоспаление и элиминацию синапсов. Активированная микроглия действует на центральные звенья восприятия боли после праймирования рецепторов врожденного иммунитета (TLR2/4, P2X, P2Y, CX3CR1), цитокиновых (ИЛ-1R, ФНО-R) и хемокиновых (CCR2, CXCR3) рецепторов. В результате усиливается синтез цитокинов (ИЛ-1-бета, ФНО-альфа, ИЛ-6) и медиаторов (простагландинов, оксидантов), активируется сигнальный путь NF-kB, изменяется уровень нейротрофического фактора мозга (BDNF). При этом количество BDNF вначале повышается как нейропротекторная реакция микроглии, но при длительном стрессе снижается за счет изменения преобладающего фенотипа на нейротоксичный. Увеличение активности рецепторов N-метил-D-аспартата (NMDA) и альфа-амино-3-гидрокси-5-метил-4-изоксазолпропионовой кислоты (AMPA) также приводит к постсинаптической гипервозбудимости, которая способствует нисходящему усилению боли [22, 23].
В ответ на стресс микроглия активируется во множестве областей мозга, включая префронтальную кору, латеральные, базолатеральные, центральные и базомедиальные ядра миндалины, зубчатую извилину гиппокампа. В гипоталамусе и миндалине микроглиальная активация приводит к секреции кортикотропин-рилизинг-гормона, в гиппокампе угнетает нейрогенез, в префронтальной коре нарушает когнитивные функции [24].
По данным ПЭТ, денервация конечности вызывает активацию глии за пределами первичной проекционной зоны, например в таламусе человека [24].
Терапевтические подходы
Рассмотрим терапевтические подходы – от общих перспективных методов купирования нейровоспаления и патологической активации микроглии до непосредственно исследуемых в настоящее время препаратов, предположительно эффективных в отношении конкретного заболевания.
Лечение рассеянного склероза
Терапевтические методы при РС, направленные на модуляцию работы микроглии, наиболее изучены по сравнению с подходами при других заболеваниях. Возможно, поэтому нейроиммуномодуляция микроглии при лечении РС может стать первым шагом на пути к широкому применению данного терапевтического принципа.
Вклад микроглии в прогрессирование РС частично объясняется ее способностью поддерживать компартментализированное воспаление. Это делает микроглию ключевой мишенью при терапии прогрессирующих форм заболевания, которые слабо отвечают на лечение, направленное преимущественно на клетки периферической иммунной системы.
Одним из перспективных методов считается ингибирование тирозинкиназы Брутона (BTK), поскольку некоторые BTK-ингибиторы способны пересекать гематоэнцефалический барьер и подавлять патогенные функции микроглии, одновременно способствуя ремиелинизации. Генетические варианты, связанные с риском развития РС, например MERTK (MER proto-oncogene, tyrosine kinase) – ген, кодирующий рецептор тирозинкиназы, экспрессируемый в микроглии, – вовлечены в процессы фагоцитоза и ремиелинации, что указывает на них как на потенциальные мишени терапевтического воздействия [25].
Понимание механизмов перехода микроглии от защитного к разрушительному состоянию в будущем может привести к созданию методов, направленных на регуляцию конкретных фенотипов микроглии [6].
Лечение болезни Паркинсона
Переход микроглии из покоящегося состояния в активное регулируется внутриклеточным Ca2+. Поэтому изменение Ca2+-сигналинга рассматривается как инструмент влияния на микроглиальный фенотип. Антагонисты кальциевых каналов L-типа (LTCC) уменьшают высвобождение провоспалительных цитокинов и ROS, что делает их потенциальными средствами снижения хронического нейровоспаления при БП и БА.
Существует устойчивая ассоциация между мутациями LRRK2 и повышенным риском развития БП. Микроглия с мутацией LRRK2-G2019S демонстрирует усиленную активацию, повышенный фагоцитоз и увеличение секреции провоспалительных цитокинов. Подавление активности LRRK2 улучшает митохондриальную функцию, снижает воспалительный ответ и повреждение дофаминергических нейронов [26].
Ингибиторы CSF1R, например PLX5622, временно уменьшают число активированных микроглиальных клеток [27]. В моделях нейродегенерации, включая БП, такая деактивация способствует сохранению дофаминергических нейронов.
В модели БП стимуляция блуждающего нерва снижает экспрессию глиальных маркеров в черной субстанции и голубом пятне, демонстрируя потенциал нейромодуляции [28].
Лечение болезни Альцгеймера
Интерес к инновационным противовоспалительным и нейропротективным иммуномодуляторным стратегиям лечения БА возрастает. Одним из перспективных направлений считается терапевтическая модуляция микроглии.
Агонисты TREM2 стимулируют пролиферацию, фагоцитоз микроглии и иммунорегуляторные программы к TREM2 с выраженным доклиническим эффектом. Усиление сигнального пути TREM2 с помощью 4D9-моноклонального агонистического антитела приводит к улучшению выживаемости макрофагов, повышению фагоцитарной активности первичной микроглии, снижению амилоидной нагрузки в мышиной модели БА, а также к ускоренному восстановлению в моделях демиелинизации.
AL002 – гуманизированное моноклональное IgG1-антитело, специально спроектированное как агонист TREM2. По результатам второй фазы клинических исследований AL002 не продемонстрировал статистически значимого замедления клинического ухудшения по сравнению с плацебо [29].
Одним из перспективных способов воздействия на микроглию является ингибирование NLRP3. MCC950 (CRID3) непосредственно ингибирует сигнально-активированную форму NLRP3, что препятствует активации каспазы 1 и выделению ИЛ-1-бета/ИЛ-18 при воспалительных стимулах. В ряде доклинических моделей нейродегенерации MCC950 снижал маркеры нейровоспаления, защищал нейроны от пироптоза и улучшал поведенческие и когнитивные показатели [30].
Антагонисты P2X7-рецептора блокируют АТФ-зависимую активацию этого ионного канала на микроглии, предотвращая NLRP3-опосредованную активацию, высвобождение ИЛ-1-бета и другие провоспалительные реакции, что приводит к снижению микроглиальной активации [1].
В ЦНС при аутоиммунных и нейродегенеративных заболеваниях, включая РС и БА, наблюдается отложение фибрина. Поэтому одним из потенциальных терапевтических подходов считается применение моноклональных антител, связывающихся с определенным эпитопом фибрина. Такой подход позволяет подавлять воспаление и окислительный стресс, индуцированные фибрином, в отсутствие влияния на систему свертывания крови [15].
В исследованиях выявлено повышение экспрессии calhm2 (calcium homeostasis modulator protein 2) в мышиной модели БА. Полное или микроглиально-специфическое удаление calhm2 приводило к значительному снижению отложения бета-амилоида, нейровоспаления и когнитивных нарушений. Механизм заключался в подавлении провоспалительной активности микроглии и усилении фагоцитоза, что восстанавливало баланс между воспалением и клиренсом амилоида [31]. Разработан селективный блокатор кальций-активируемого калиевого канала KCa3.1, экспрессированного в микроглии, – сеникапок. Ингибирование KCa3.1 снижает провоспалительную активацию микроглии в моделях БА [32].
Лечение инсульта
В настоящее время изучаются различные методы иммунотерапии инсульта, в том числе модулирующие активность микроглии. Наиболее перспективными терапевтическими мишенями являются каспаза 1 и каспаза 3, NLRP1-, NLRP3-белки.
Исследователи оценивали эффективность ингибитора каспазы 1 в первичных кортикальных нейронах в условиях ишемии. Применяли возрастающие концентрации ингибитора каспазы 1 Ac-YVAD-CMK, после чего анализировали уровни расщепленной каспазы 1. Концентрации ингибитора каспазы 1 выше 30 мкМ снижали уровни расщепленной каспазы 1 и каспазы 3 – маркера апоптоза [33].
Достаточно давно установлено, что внутривенный иммуноглобулин (IVIg) значительно снижает уровни NLRP1, NLRP3, ASC, XIAP, каспазы 1, каспазы 11, ИЛ-1-бета и ИЛ-18, но молекулярный механизм этого эффекта неизвестен. Исследование, проведенное на модели церебральной ишемии, подтвердило наблюдаемые последствия. Установлено, что повышение экспрессии белков инфламмасомы NLRP1 и NLRP3 и предшественников ИЛ-1-бета и ИЛ-18 в нейронах при ишемии может быть индуцировано активацией рецепторов распознавания образов (PRRs), таких как TLR, рецептор продвинутых продуктов гликирования и ИЛ-1R1, которые распознают DAMP-сигналы, высвобождаемые в инфарктном очаге. Сигналинг через TLR приводит к праймингу микроглии [33].
Стимуляция блуждающего нерва на похожих моделях подавляет пироптоз, опосредованный инфламмасомой NLRP3, и снижает уровни провоспалительных цитокинов ИЛ-1-бета и ИЛ-18 в ткани мозга, что демонстрирует нейропротективный эффект, частично обусловленный подавлением микроглиального воспаления [20].
Лечение хронической боли
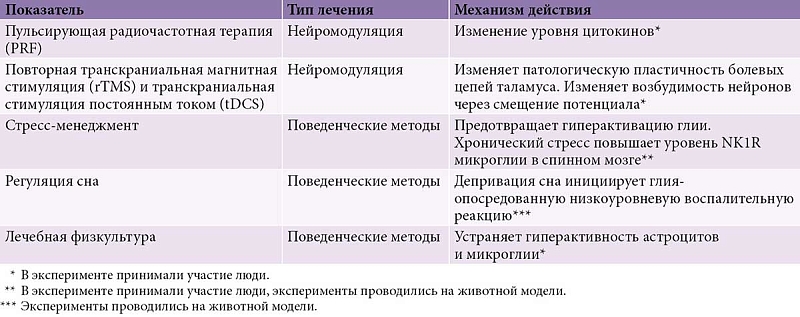
Модуляция активности микроглии рассматривается как перспективная стратегия для купирования хронической боли, стресс-индуцированной гипералгезии, фибромиалгии и висцеральной гипералгезии. В настоящее время активно исследуются методы нейромодуляции – транскраниальная магнитная стимуляция и пульсирующая радиочастотная терапия (табл. 1), а также разнообразные фармакологические подходы (табл. 2). За счет постсинаптической гипервозбудимости хронический стресс приводит к активации микроглии спинного мозга и тем самым способствует развитию нейровоспаления. В связи с этим рассматривается применение поведенческих терапевтических вмешательств, которые также могут косвенно влиять на фенотипический профиль иммунных медиаторов, выделяемых микроглией [24].
Заключение
Комплексное рассмотрение новых подходов к фенотипическому разделению микроглии, нейроиммунным взаимодействиям, ключевым путям ее активации и возможностям терапевтической модуляции при различных заболеваниях позволяет глубже понять клиническую значимость микроглиальных механизмов и сформировать основу для разработки инновационных нейроиммуномодулирующих стратегий лечения заболеваний нервной системы. Успех первых доклинических и клинических исследований подтверждает перспективность данного направления, одновременно подчеркивая необходимость разработки более фенотип-ориентированных методов и расширения доказательной базы за счет дальнейших клинических исследований.
A.A. Shumkina
I.M. Sechenov First Moscow State Medical University
I.I. Mechnikov Scientific Research Institute of Vaccines and Serums, Moscow
Contact person: Anna A. Shumkina, shumkina_a_a@student.sechenov.ru
Microglia are key regulators of neuroinflammation and are involved in the pathogenesis of multiple sclerosis, Parkinson's disease, Alzheimer's disease, stroke, chronic pain, and other diseases. The review presents microglia phenotypes, signaling activation pathways, and disease-specific mechanisms. Modern approaches to therapeutic modulation of microglia and prospects for its use in clinical neurology are considered.



Уважаемый посетитель uMEDp!
Уведомляем Вас о том, что здесь содержится информация, предназначенная исключительно для специалистов здравоохранения.
Если Вы не являетесь специалистом здравоохранения, администрация не несет ответственности за возможные отрицательные последствия, возникшие в результате самостоятельного использования Вами информации с портала без предварительной консультации с врачом.
Нажимая на кнопку «Войти», Вы подтверждаете, что являетесь врачом или студентом медицинского вуза.