Высокая эффективность и безопасность инсулинотерапии возможны
- Аннотация
- Статья
- Ссылки
- English


В последние десятилетия рост заболеваемости сахарным диабетом (СД) сравнивают с неинфекционной пандемией. Так, в Российской Федерации за 20 лет число пациентов увеличилось в 2,5 раза [1].
При СД лидирующими осложнениями являются сердечно-сосудистые. Они же остаются главной причиной смерти пациентов с СД 2 типа. Установлено, что около 68% лиц старше 65 лет умирают от заболеваний сердца, 16% – от инсульта [2, 3].
Сахарный диабет часто сочетается с такими нарушениями, как артериальная гипертензия и дислипидемия, что в совокупности обусловливает повышенный сердечно-сосудистый риск, раннее развитие заболеваний сердечно-сосудистой системы, а также серьезных осложнений, нередко с фатальным исходом. Наиболее часто СД ассоциируется с ишемической болезнью сердца, инсультом, облитерирующим атеросклерозом сосудов нижних конечностей, кардиомиопатией, сердечной недостаточностью [4]. Увеличение сердечно-сосудистой смертности при СД можно объяснить несколькими причинами: гипергликемией, гипогликемией, снижением парасимпатической активности вегетативной нервной системы. При проведении метаанализа базы данных MEDLINE за 31 год E. Barkaudah и соавт. установили, что более высокие показатели смертности также ассоциировались с более высоким артериальным давлением [5].
Связь между гипергликемией и сердечно-сосудистыми заболеваниями была доказана в исследовании DECODE [6]. Так, гликемия более 8 ммоль/л увеличивала риск развития сердечно-сосудистой патологии в два раза. Снижение этого показателя на 2 ммоль/л уменьшало риск смертельного исхода на 20–30%.
Уровень постпрандиальной глюкозы в крови, так же как уровень глюкозы плазмы натощак и гликированного гемоглобина (HbAlc), признан достоверным предиктором развития микрососудистых осложнений СД. В ряде исследований также отмечена его роль в развитии макрососудистых осложнений [7, 8].
Задолго до гипергликемии обнаруживается такой метаболический дефект, как инсулинорезистентность [9]. Инсулинорезистентность считается ключевым звеном не только нарушения толерантности к глюкозе или СД 2 типа, но и абдоминального ожирения, артериальной гипертензии, атерогенной дислипидемии, что обозначается как метаболический синдром [10].
Инсулинорезистентность в печени и жировой ткани сопровождается гиперпродукцией липопротеинов низкой плотности, нарушением катаболизма атерогенных липопротеиновых остатков (ремнантных частиц) и гиперкатаболизмом липопротеинов высокой плотности, что ведет к развитию атерогенной дислипидемии. Для последней характерно повышение уровня богатых триглицеридами липопротеинов (очень низкой плотности, промежуточной плотности), уменьшение содержания липопротеинов высокой плотности и увеличение – липопротеинов низкой плотности [11].
Утилизация глюкозы миокардом при СД значительно снижается, приводя к активации β-окисления свободных жирных кислот. Причиной тому служит уменьшение активности глюкозных транспортеров 1 и 4. Повышенное поступление свободных жирных кислот в кардиомиоциты превосходит возможность их окисления, что сопровождается накоплением триглицеридов в миокарде, его сократительной дисфункцией и гипертрофией [12]. Свободные жирные кислоты ингибируют пируватдегидрогеназу, что нарушает энергетический обмен в миокарде и ведет к накоплению промежуточных продуктов гликолиза, воспалительных цитокинов, церамидов и усилению апоптоза [13]. Оксидативный стресс, тканевая гипоксия из-за разрежения микроциркуляторного русла, инфильтрация провоспалительными иммунокомпетентными клетками, накопление конечных продуктов гликозилирования и липотоксичность приводят к гибели кардиомиоцитов с исходом в заместительный фиброз [14]. Кроме того, гипергликемия вызывает активацию протеинкиназы С в фибробластах, что обусловливает увеличение продукции и отложения коллагена [15].
Эндотелиальная дисфункция, присущая СД уже на ранних стадиях, характеризуется снижением биодоступности оксида азота (NO) и сопутствующим повышением образования супероксид-аниона. Потеря биодоступности NO предшествует развитию атеросклероза и служит независимым фактором неблагоприятных сердечно-сосудистых событий [16]. В настоящее время известно несколько путей влияния избытка свободных радикалов на эндотелиальную функцию:
- супероксид-анион быстро инактивирует NO и превращает его в пероксинитрит – мощный оксидант, который легко проникает через фосфолипидные мембраны и вызывает нитрование субстратов, блокируя регуляторные рецепторы, ферменты-инактиваторы свободных радикалов и ключевые кофакторы эндотелиальной NO-синтазы;
- митохондриальная продукция супероксид-аниона повышает внутриклеточное образование продуктов конечного гликозилирования, что усиливает образование свободных радикалов и провоспалительных цитокинов в клетках сосудов, а также эндотелиальную экспрессию различных молекул адгезии, участвующих в атерогенезе;
- активация рецепторов продуктов конечного гликозилирования повышает внутриклеточное образование супероксид-аниона, что играет важную роль в развитии атеросклеротического поражения.
Накопление конечных продуктов гликозилирования при СД приводит к микрососудистому ремоделированию с утолщением базальной мембраны и образованием микроаневризм. Эти структурные изменения сопровождаются развитием эндотелиальной дисфункции, снижением плотности сосудов и повышением их проницаемости [17].
Инсулинорезистентность независимо от других факторов значительно увеличивает вероятность развития сердечно-сосудистой патологии. При данном нарушении отмечается более высокая частота множественного атеросклеротического поражения коронарных сосудов, чем при сохраненной чувствительности к инсулину [18].
Атеросклероз и инсулинорезистентность имеют схожие патофизиологические механизмы, главным образом за счет действия двух главных провоспалительных цитокинов: фактора некроза опухолей α и интерлейкина 6 [19].
Повышение уровня маркеров воспалительной реакции, эндотелиальной дисфункции признаны факторами высокого риска развития острых атеротромботических событий [20].
Степень активности системного воспаления у пациентов с ишемической болезнью сердца и СД можно рассматривать как наиболее важную характеристику ускорения повреждения сосудистой стенки и деструктивных изменений в атеросклеротических бляшках.
Важность воспаления в развитии и прогрессировании атеросклероза доказана в ряде исследований. Так, установлена связь между маркерами воспаления, показателями ремоделирования сердечно-сосудистой системы и тяжестью атеросклероза [21, 22].
Сахарный диабет 2 типа ассоциируется с провоспалительным иммунным статусом и увеличением уровня циркулирующих маркеров воспаления, в частности фибриногена, интерлейкинов 1 и 6, высокочувствительного С-реактивного белка, фактора некроза опухолей α [23–25].
У больных СД атеросклероз коронарных артерий развивается в более молодом возрасте и поражает дистальные сегменты коронарных артерий. Кроме того, у таких пациентов коллатеральное кровообращение развивается хуже, что может быть вызвано нарушением продукции или ответа на сосудистые факторы роста [26]. Резерв коронарного кровотока у них также снижен, даже без обструктивного атеросклероза коронарных артерий. Это объясняется тем, что гипергликемия увеличивает синтез вазоконстрикторных простагландинов эндотелием и активирует протеинкиназу С [27].
Независимым фактором риска развития сердечно-сосудистых заболеваний также является ожирение [28]. В настоящее время жировая ткань рассматривается как эндокринный орган, в котором вырабатываются адипокины, такие как лептин, адипонектин, резистин, оментин, висфатин. Адипокины участвуют в системном воспалении, развитии инсулинорезистентности, атерогенной дислипидемии и СД 2 типа. В частности, уровень лептина положительно коррелировал с содержанием триглицеридов и свободных жирных кислот у лиц с СД 2 типа и ожирением [29]. Это в свою очередь свидетельствовало о связи лептина с липотоксичностью.
Дислипидемия и ожирение ассоциируются с атеросклерозом, а последний – с ишемической болезнью сердца.
Хроническая гипергликемия непосредственно влияет на структуру и функцию миокарда [14]. Общепризнанного определения диабетической кардиомиопатии не существует. Часто она характеризуется как нарушение структуры или функции миокарда без поражения эпикардиальных коронарных артерий, артериальной гипертензии и значимых клапанных пороков [27]. В настоящее время описаны структурные и функциональные различия между двумя фенотипами диабетической кардиомиопатии: рестриктивным и дилатационным. Так, при рестриктивном фенотипе развивается гипертрофия кардиомиоцитов с сохранением нормальной структуры саркомеров и реактивным фиброзом. При дилатационном фенотипе повреждение кардиомиоцитов сопровождается утратой саркомеров, повышается количество межклеточного коллагена, развивается заместительный фиброз. При обоих фенотипах наблюдается редукция микроциркуляторного русла и отложение в нем конечных продуктов гликозилирования. Среди патогенетических механизмов диабетической кардиомиопатии особая роль отводится нарушению метаболизма свободных жирных кислот с развитием липотоксичности, ускорению апоптоза, автономной невропатии, микрососудистым поражениям и эндотелиальной дисфункции на фоне инсулинорезистентности и гиперинсулинемии [14].
Сахарный диабет 2 типа также является независимым фактором риска развития сердечной недостаточности. Еще в исследовании UKPDS были получены данные об увеличении риска ее развития на 16% при возрастании уровня HbA1с на 1% [8]. Согласно результатам Фремингемского исследования, наличие СД ассоциировалось с увеличением риска развития сердечной недостаточности в два раза у мужчин и пять раз у женщин [30]. В популяционном исследовании, включавшем 1,9 млн пациентов с СД 2 типа без явных признаков сердечно-сосудистых заболеваний, за пять с половиной лет наблюдения частота возникновения сердечной недостаточности оказалась выше, чем частота инфаркта миокарда или инсульта [31]. Риск развития сердечной недостаточности после перенесенного инфаркта миокарда у больных СД увеличивался в два-три раза [32]. Прогноз у пациентов с сердечной недостаточностью и СД был хуже, чем у лиц без СД.
D.S.H. Bell рассматривал сердечную недостаточность как частое, забытое и зачастую фатальное осложнение СД [33].
В основе патогенеза сердечной недостаточности при СД лежит кардиотоксическая тетрада: ишемическая болезнь сердца, артериальная гипертензия, диабетическая кардиомиопатия и увеличение объема внеклеточной жидкости [33].
Сахарный диабет 2 типа приводит к развитию сердечной недостаточности через механизмы как опосредованные атеросклерозом, так и не зависящие от него. В связи с этим выделяют два фенотипа сердечной недостаточности – со сниженной и сохранной фракцией выброса. Сердечная недостаточность со сниженной фракцией выброса рассматривается как следствие прямого повреждения миокарда (некроз, апоптоз) в результате ишемии и других факторов, сердечная недостаточность с сохранной фракцией выброса – как системное заболевание, характеризующееся воспалением, микрососудистой и эндотелиальной дисфункцией [34].
Важную роль в развитии сердечно-сосудистых заболеваний при СД играет кардиальная автономная невропатия. Поражение блуждающего нерва приводит к относительному преобладанию симпатической активности и активации ренин-ангиотензин-альдостероновой системы, что сопровождается увеличением частоты сердечных сокращений, ударного объема, периферического сосудистого сопротивления, задержкой натрия и воды. Как следствие, развивается дисфункция миокарда левого желудочка [35].
Микроальбуминурия не только ранний маркер развития диабетической нефропатии, но и фактор риска возникновения сердечно-сосудистого заболевания [36]. Диабетическая нефропатия характеризуется увеличением задержки натрия [37].
Согласно рекомендациям экспертов Европейского общества кардиологов [38], лица с СД и сердечно-сосудистыми заболеваниями или с СД и поражением органов-мишеней, такими как протеинурия или почечная недостаточность (расчетная скорость клубочковой фильтрации менее 30 мл/мин/1,73 м2), относятся к группе очень высокого риска (десятилетний риск фатального исхода от сердечно-сосудистого заболевания превышает 10%). Пациенты с СД и тремя основными факторами риска и более (возраст, артериальная гипертензия, дислипидемия, курение, ожирение) или с продолжительностью СД более 20 лет также относятся к группе очень высокого риска.
Профилактика развития перечисленных диабетических осложнений требует проведения своевременной и эффективной сахароснижающей терапии. Несмотря на значительные достижения в лечении СД 2 типа, контроль заболевания нельзя признать оптимальным. Это в свою очередь приводит к длительному воздействию повышенного уровня глюкозы на организм.
В исследовании J.B. Brown и соавт. показано, что на момент инициации инсулинотерапии в среднем пациенты имели в течение десяти лет уровень HbA1c более 7%, в течение пяти лет – более 8% (рис. 1) [39].
В российском исследовании A1chieve показано, что при инициации базальной инсулинотерапии средний уровень HbA1c у пациентов был равен 9,7%, готовыми смесями инсулинов – 10,1%, базис-болюсной терапии – 10,4% [40]. Полученные данные свидетельствуют о тенденции начинать инсулинотерапию только при уровне HbA1c более 9%. Такая клиническая пассивность/инертность приводит к неудовлетворительным результатам в отношении достижения целевых значений гликемии даже при возможности применения высокоэффективных препаратов.
Аналоги базального инсулина пролонгированного действия внесли существенный вклад в лечение больных с СД в связи с более длительным и значительно более ровным профилем действия [41]. Благодаря особенностям препаратов нового поколения удалось уменьшить частоту гипогликемий. Как следствие, началось их широкое внедрение в клиническую практику.
При постановке диагноза СД 2 типа необходимо предупреждать пациентов, что инсулинотерапия неизбежна из-за значительного снижения функции β-клеток и очень важно, чтобы переход на нее произошел своевременно. Основное преимущество инсулинотерапии заключается в компенсации недостатка эндогенной секреции инсулина и, как следствие, прогрессирующей дисфункции β-клеток.
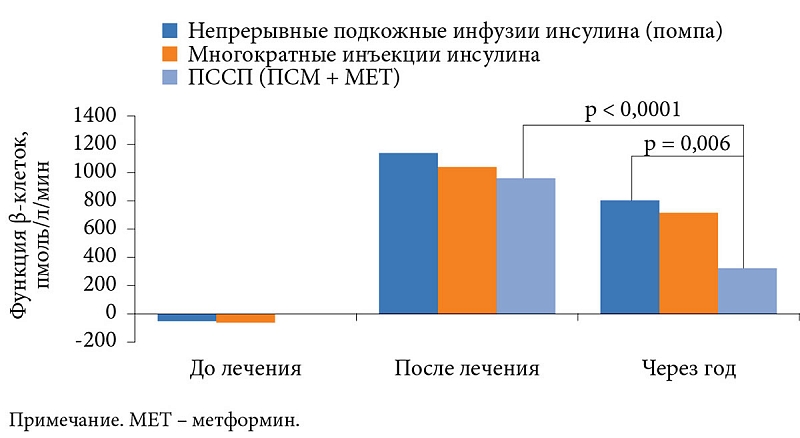
Влияние раннего начала инсулинотерапии на функцию β-клеток изучалось в рандомизированном исследовании, в которое были включены 382 пациента с впервые выявленным СД 2 типа, индексом массы тела 25 кг/м2. Средний их возраст составил 51 года. На момент включения в исследование уровень HbA1c в среднем достигал 9,7%.
Больные были рандомизированы на несколько групп терапии: непрерывной подкожной инфузии инсулина, ежедневных инъекций инсулина или приема пероральных сахароснижающих препаратов (ПССП). Нормогликемия поддерживалась в течение двух недель [42]. Результаты исследования свидетельствовали о восстановлении функции β-клеток у получавших инсулин в отличие от применявших ПССП (рис. 2).
В настоящее время в Российской Федерации применяются два инновационных инсулина второго поколения – гларгин 300 ЕД (Туджео СолоСтар®) и деглудек (Тресиба®). В отличие от гларгина 100 ЕД/мл концентрация активного вещества в Туджео СолоСтар® составляет 300 ЕД/мл, то есть в три раза выше на 1 мл раствора. Поэтому при введении одинакового количества единиц объем гларгина 300 ЕД/мл соответствует одной трети объема гларгина 100 ЕД/мл. В результате уменьшения площади поверхности преципитата скорость высвобождения гларгина 300 ЕД/мл меньше, чем гларгина 100 ЕД/мл [43]. Следует отметить, что именно этим обусловлены лучшие фармакокинетические и фармакодинамические профили инсулина гларгин 300 ЕД/мл.
Клиническая эффективность и безопасность инсулина гларгин 300 ЕД/мл и инсулина гларгин 100 ЕД/мл оценены в исследовании EDITION. Туджео СолоСтар® продемонстрировал явное преимущество перед Лантусом [44]. Результаты трех исследований EDITION показали сопоставимость эффектов гларгина 300 и 100 ЕД/мл в отношении показателя «достижение гликемического контроля в течение шести месяцев». Тяжелая гипогликемия на фоне применения обоих препаратов встречалась редко, однако преимущество сохранялось за гларгином 300 ЕД/мл. Разница между препаратами в отношении кумулятивного количества случаев ночной подтвержденной или тяжелой гипогликемии составила 14%. Это можно объяснить более длительным депонированием гларгина 300 ЕД/мл в подкожной жировой клетчатке и увеличением ферментативной инактивации тканевыми пептидазами в месте инъекции.
Особый интерес представляют результаты исследования BRIGHT.
BRIGHT – первое прямое сравнительное исследование клинической эффективности и безопасности гларгина 300 ЕД/мл и деглудека 100 ЕД/мл. В 24-недельном исследовании приняли участие пациенты с неконтролируемым СД 2 типа, ранее не получавшие инсулин. Больные были рандомизированы в соотношении 1:1 в группу гларгина 300 ЕД/мл (n = 466) или группу деглудека 100 ЕД/мл (n = 463).
Оба препарата вводили в вечернее время.
Дозу инсулина подбирали до достижения уровня глюкозы плазмы натощак 4,4–5,6 ммоль/л при самостоятельном измерении пациентом.
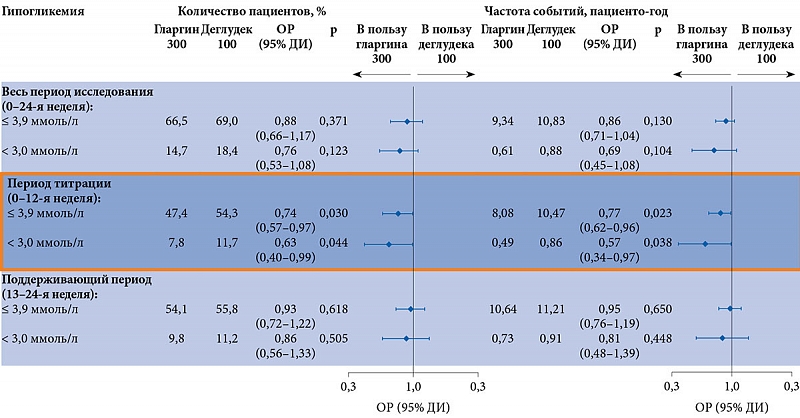
Первичной конечной точкой служила динамика НbА1с к концу 24-й недели. В конце исследования в обеих группах наблюдалось сопоставимое снижение показателей HbА1с по сравнению с исходными – с 8,7% в группе гларгина 300 ЕД/мл и 8,6% в группе деглудека 100 ЕД/мл до 7,0%. Наименьшие квадраты средних различий составили 0,05% (95%-ный доверительный интервал (ДИ) -0,15–0,05), что свидетельствовало о не меньшей эффективности гларгина, чем деглудека (р < 0,0001). Доля пациентов с эпизодами гипогликемий, зафиксированными в ходе исследования, и частота эпизодов гипогликемий через 24 недели были также сопоставимы в обеих группах. Однако в течение периода активной титрации дозы (с начала терапии и до 12-й недели) доля пациентов с подтвержденной гипогликемией и частота эпизодов подтвержденной гипогликемии (≤ 3,9 и < 3,0 ммоль/л) в любое время суток были ниже в группе гларгина 300 ЕД/мл [45].
Таким образом, одного из самых значимых барьеров для старта инсулинотерапии можно избежать с помощью инициации длительно действующих аналогов базального инсулина второго поколения.
В отношении достижения целевой гликемии ключевым является подбор дозы инсулина в течение 12 недель. Именно в этом промежутке титрации желательно достичь необходимых доз инсулина и целевых показателей глюкозы в крови.
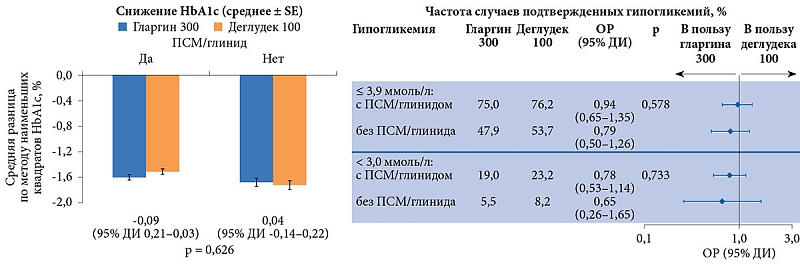
В исследовании BRIGHT в обеих группах титрация дозы инсулина была эффективной. В целом в каждой группе 47% достигли целевого уровня HbA1c < 7,0% (рис. 3). Однако доля достигших этого показателя без гипогликемий к 12-й неделе была выше среди получавших гларгин 300 ЕД/мл (рис. 4).
Подтвержденная гипогликемия включала задокументированную симптоматическую или бессимптомную гипогликемию (≤ 3,9 или < 3,0 ммоль/л) или тяжелые события, если они имели место. Только одна пациентка перенесла тяжелую гипогликемию (одно событие) в группе гларгина 300 ЕД/мл из-за пропуска вечернего приема пищи без соответствующей коррекции дозы инсулина. Это случилось после нетяжелого эпизода двумя днями ранее.
В течение 24 недель в группах гларгина 300 ЕД/мл и деглудека 100 ЕД/мл наблюдались сопоставимые гликемический контроль и частота гипогликемий независимо от использования на скрининге таких секретогенов эндогенного инсулина, как производные сульфонилмочевины (ПСМ) и глиниды (рис. 5).
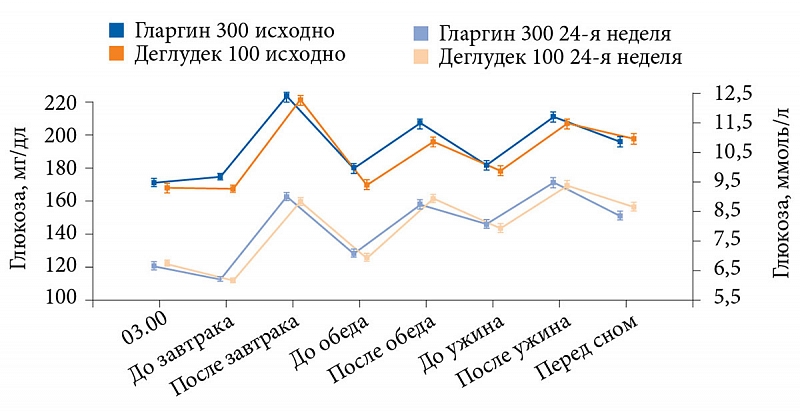
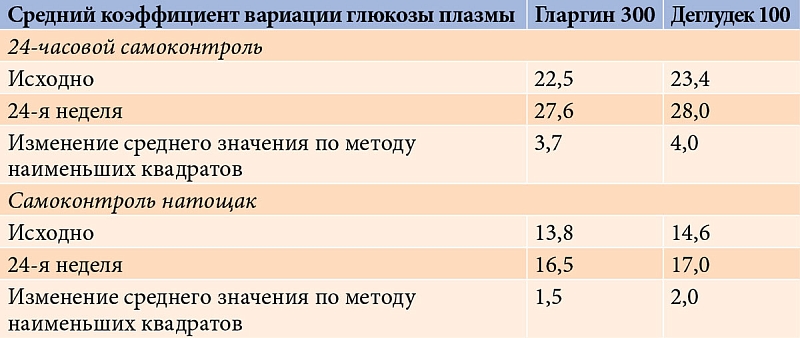
Полученные результаты позволили исследователям сделать вывод, что между гларгином 300 ЕД/мл и деглудеком 100 ЕД/мл больше сходства, чем различий [45]. Это подтверждалось и сопоставимым восьмиточечным профилем самоконтроля глюкозы в крови и профилем вариабельности (рис. 6 и табл. 1).
Как видно на рис. 6 и в табл. 1, через 24 недели в двух группах анализируемые профили были сопоставимы.
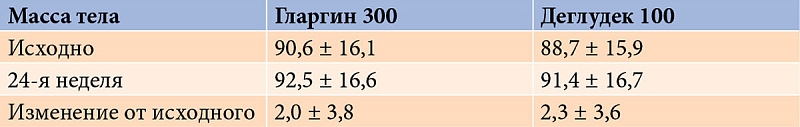
Аналогичная динамика в обеих группах зафиксирована и в отношении массы тела (табл. 2).
Таким образом, аналоги пролонгированного инсулина второго поколения имеют ряд преимуществ. Так, увеличение концентрации гларгина до 300 ЕД/мл позволило обеспечить более длительное действие и более ровный суточный профиль, имитирующий физиологическую секрецию базального инсулина. Как следствие, повысились эффективность и безопасность лечения препаратом.
Результаты прямого сравнительного исследования BRIGHT по оценке эффективности и безопасности инновационных аналогов базального инсулина пролонгированного действия второго поколения свидетельствуют о сходном снижении уровня HbA1c на фоне применения гларгина 300 ЕД/мл и деглудека 100 ЕД/мл. Однако доля пациентов с подтвержденной гипогликемией и частота эпизодов подтвержденной гипогликемии в любое время суток были ниже в группе гларгина 300 ЕД/мл [45].
Как следствие, применение гларгина 300 ЕД/мл может позволить избежать одного из самых значимых барьеров для старта инсулинотерапии.
A.M. Mkrtumyan, MD, PhD, Prof., D.V. Kileynikov, PhD
A.I. Yevdokimov Moscow State University of Medicine and Dentistry
Contact person: Ashot M. Mkrtumyan, vagrashot@mail.ru
Diabetes mellitus is characterized by chronic hyperglycemia and insulin resistance of varying degrees. In addition, this pathology is often combined with disorders such as hypertension and dyslipidemia. In combination, this increases the cardiovascular risk and, as a result, contributes to the early development of diseases of the cardiovascular system. Achieving targeted glycemic control is associated with the reduced risk of late complications developing of diabetes mellitus. However, many patients require insulin therapy to maintain adequate control. Timely administration of insulin can prevent the progression of diabetes, reduce the risk of its complications and have less pronounced side effects. Basal insulin is the preferred treatment option in most cases of non-achievement of the target glycemia. Longer-lasting effects, reduced blood glucose variability and the risk of hypoglycemia observed with the use of basal insulin analogues of the latest generation simplify dose titration and can increase patient adherence to treatment.
Уважаемый посетитель uMEDp!
Уведомляем Вас о том, что здесь содержится информация, предназначенная исключительно для специалистов здравоохранения.
Если Вы не являетесь специалистом здравоохранения, администрация не несет ответственности за возможные отрицательные последствия, возникшие в результате самостоятельного использования Вами информации с портала без предварительной консультации с врачом.
Нажимая на кнопку «Войти», Вы подтверждаете, что являетесь врачом или студентом медицинского вуза.