Cerebrum diabeticum: —Б—Г—Й–µ—Б—В–≤—Г–µ—В –ї–Є –і–Є–∞–±–µ—В–Є—З–µ—Б–Ї–∞—П —Н–љ—Ж–µ—Д–∞–ї–Њ–њ–∞—В–Є—П?
- –Р–љ–љ–Њ—В–∞—Ж–Є—П
- –°—В–∞—В—М—П
- –°—Б—Л–ї–Ї–Є
- English
–°—Г–±—Б—В—А–∞—В —Н–љ—Ж–µ—Д–∞–ї–Њ–њ–∞—В–Є–Є –Њ—Б—В–∞–µ—В—Б—П –љ–µ—П—Б–љ—Л–Љ, –∞ –µ–µ —Б—В–∞—В—Г—Б, –Ї—А–Є—В–µ—А–Є–Є –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–Є –њ—А–Њ–і–Њ–ї–ґ–∞—О—В –Њ–±—Б—Г–ґ–і–∞—В—М—Б—П, –≤ —Б–≤—П–Ј–Є —Б —З–µ–Љ –≤ –њ–Њ—Б–ї–µ–і–љ–Є–µ –≥–Њ–і—Л –і–∞–љ–љ—Л–є —В–µ—А–Љ–Є–љ –њ—А–Є–Љ–µ–љ—П–µ—В—Б—П —Б—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ —А–µ–ґ–µ. –Я—А–Є –°–Ф 1 —В–Є–њ–∞ –Ф–≠ –љ–Њ—Б–Є—В –њ–µ—А–≤–Є—З–љ—Л–є —Е–∞—А–∞–Ї—В–µ—А –Є —Б–≤—П–Ј–∞–љ–∞ —Б –љ–∞—А—Г—И–µ–љ–Є–µ–Љ –і–µ–є—Б—В–≤–Є—П –Є–љ—Б—Г–ї–Є–љ–∞ –Є –≥–Є–њ–µ—А–≥–ї–Є–Ї–µ–Љ–Є–µ–є, —В–Њ–≥–і–∞ –Ї–∞–Ї –њ—А–Є –°–Ф 2 —В–Є–њ–∞ –≤ –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ–є —З–∞—Б—В–Є —Б–ї—Г—З–∞–µ–≤ –Њ–љ–∞ —П–≤–ї—П–µ—В—Б—П —Б–ї–µ–і—Б—В–≤–Є–µ–Љ —Б–Њ—Б—Г–і–Є—Б—В—Л—Е –Њ—Б–ї–Њ–ґ–љ–µ–љ–Є–є.
–°—Г–±—Б—В—А–∞—В —Н–љ—Ж–µ—Д–∞–ї–Њ–њ–∞—В–Є–Є –Њ—Б—В–∞–µ—В—Б—П –љ–µ—П—Б–љ—Л–Љ, –∞ –µ–µ —Б—В–∞—В—Г—Б, –Ї—А–Є—В–µ—А–Є–Є –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–Є –њ—А–Њ–і–Њ–ї–ґ–∞—О—В –Њ–±—Б—Г–ґ–і–∞—В—М—Б—П, –≤ —Б–≤—П–Ј–Є —Б —З–µ–Љ –≤ –њ–Њ—Б–ї–µ–і–љ–Є–µ –≥–Њ–і—Л –і–∞–љ–љ—Л–є —В–µ—А–Љ–Є–љ –њ—А–Є–Љ–µ–љ—П–µ—В—Б—П —Б—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ —А–µ–ґ–µ. –Я—А–Є –°–Ф 1 —В–Є–њ–∞ –Ф–≠ –љ–Њ—Б–Є—В –њ–µ—А–≤–Є—З–љ—Л–є —Е–∞—А–∞–Ї—В–µ—А –Є —Б–≤—П–Ј–∞–љ–∞ —Б –љ–∞—А—Г—И–µ–љ–Є–µ–Љ –і–µ–є—Б—В–≤–Є—П –Є–љ—Б—Г–ї–Є–љ–∞ –Є –≥–Є–њ–µ—А–≥–ї–Є–Ї–µ–Љ–Є–µ–є, —В–Њ–≥–і–∞ –Ї–∞–Ї –њ—А–Є –°–Ф 2 —В–Є–њ–∞ –≤ –Ј–љ–∞—З–Є—В–µ–ї—М–љ–Њ–є —З–∞—Б—В–Є —Б–ї—Г—З–∞–µ–≤ –Њ–љ–∞ —П–≤–ї—П–µ—В—Б—П —Б–ї–µ–і—Б—В–≤–Є–µ–Љ —Б–Њ—Б—Г–і–Є—Б—В—Л—Е –Њ—Б–ї–Њ–ґ–љ–µ–љ–Є–є.
–°–∞—Е–∞—А–љ—Л–є –і–Є–∞–±–µ—В (–°–Ф) вАУ –Њ–і–љ–Њ –Є–Ј¬†—Б–∞–Љ—Л—Е —З–∞—Б—В—Л—Е —Е—А–Њ–љ–Є—З–µ—Б–Ї–Є—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є, –њ–Њ—А–∞–ґ–∞—О—Й–µ–µ –і–Њ¬†10% –љ–∞—Б–µ–ї–µ–љ–Є—П. –Э–µ—А–≤–љ–∞—П —Б–Є—Б—В–µ–Љ–∞ вАУ –Њ–і–љ–∞ –Є–Ј¬†–Њ—Б–љ–Њ–≤–љ—Л—Е –Љ–Є—И–µ–љ–µ–є –°–Ф. –Ґ—А–∞–і–Є—Ж–Є–Њ–љ–љ–Њ —Б—З–Є—В–∞–ї–Њ—Б—М, —З—В–Њ –њ—А–Є –°–Ф –≤¬†–њ–∞—В–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–є –њ—А–Њ—Ж–µ—Б—Б –Љ–Њ–ґ–µ—В –≤–Њ–≤–ї–µ–Ї–∞—В—М—Б—П —Ж–µ–љ—В—А–∞–ї—М–љ–∞—П –љ–µ—А–≤–љ–∞—П —Б–Є—Б—В–µ–Љ–∞ (–¶–Э–°), –Њ–і–љ–∞–Ї–Њ –њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ —Б—В—А–∞–і–∞–µ—В –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–∞—П –љ–µ—А–≤–љ–∞—П —Б–Є—Б—В–µ–Љ–∞ (–Я–Э–°). –Я–µ—А–µ—Д—А–∞–Ј–Є—А—Г—П –Ј–љ–∞–Љ–µ–љ–Є—В–Њ–µ –Є–Ј—А–µ—З–µ–љ–Є–µ, –Љ–Њ–ґ–љ–Њ —Б–Ї–∞–Ј–∞—В—М, —З—В–Њ –°–Ф ¬Ђ–ї–Є–ґ–µ—В¬ї –¶–Э–°, –љ–Њ¬†¬Ђ–Ї—Г—Б–∞–µ—В¬ї –Я–Э–°.
–Т¬†–њ–Њ—Б–ї–µ–і–љ–Є–µ –і–µ—Б—П—В–Є–ї–µ—В–Є—П –њ—А–µ–і—Б—В–∞–≤–ї–µ–љ–Є—П –Є–Ј–Љ–µ–љ–Є–ї–Є—Б—М. –£—Б—В–∞–љ–Њ–≤–ї–µ–љ–Њ, —З—В–Њ –і–Є—Б—Д—Г–љ–Ї—Ж–Є—П –≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ вАУ —З–∞—Б—В–Њ–µ —П–≤–ї–µ–љ–Є–µ –њ—А–Є –°–Ф. –Ю–і–љ–∞–Ї–Њ –µ–µ –њ–∞—В–Њ–≥–µ–љ–µ–Ј, –љ–Њ–Ј–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–є —Б—В–∞—В—Г—Б, –Ї–ї–Є–љ–Є–Ї–∞, –њ—А–Њ–≥–љ–Њ–Ј, –Ї—А–Є—В–µ—А–Є–Є –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–Є, –њ–Њ–і—Е–Њ–і—Л –Ї¬†—В–µ—А–∞–њ–Є–Є –њ–ї–Њ—Е–Њ –Є–Ј—Г—З–µ–љ—Л –Є¬†–≤—Л–Ј—Л–≤–∞—О—В –і–Є—Б–Ї—Г—Б—Б–Є–Є [1вАУ3].
–Ъ–ї–∞—Б—Б–Є—Д–Є–Ї–∞—Ж–Є—П –њ–Њ—А–∞–ґ–µ–љ–Є–є –≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ –њ—А–Є¬†—Б–∞—Е–∞—А–љ–Њ–Љ¬†–і–Є–∞–±–µ—В–µ
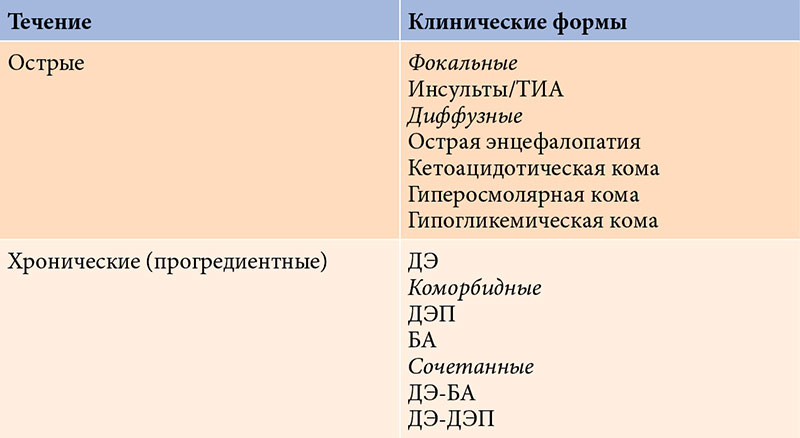
–Я–Њ—А–∞–ґ–µ–љ–Є–µ –≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ –њ—А–Є –°–Ф –Љ–Њ–ґ–µ—В –±—Л—В—М –Њ—Б—В—А—Л–Љ –Є–ї–Є —Е—А–Њ–љ–Є—З–µ—Б–Ї–Є–Љ (—Б–Љ. —В–∞–±–ї–Є—Ж—Г). –Ю—Б—В—А—Л–µ –њ–Њ—А–∞–ґ–µ–љ–Є—П –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ—Л –Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Є–Љ–Є –љ–∞—А—Г—И–µ–љ–Є—П–Љ–Є (–Њ—Б—В—А—Л–µ –Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Є–µ —Н–љ—Ж–µ—Д–∞–ї–Њ–њ–∞—В–Є–Є, –Є–ї–Є –Ї–Њ–Љ—Л: –≥–Є–њ–µ—А–≥–ї–Є–Ї–µ–Љ–Є—З–µ—Б–Ї–∞—П, –Ї–µ—В–Њ–∞—Ж–Є–і–Њ—В–Є—З–µ—Б–Ї–∞—П, –≥–Є–њ–Њ–≥–ї–Є–Ї–µ–Љ–Є—З–µ—Б–Ї–∞—П –Є¬†—В.–і.). –Ъ–∞–Ї –њ—А–∞–≤–Є–ї–Њ, –Њ–љ–Є –Њ–±—А–∞—В–Є–Љ—Л, –Њ–і–љ–∞–Ї–Њ –Љ–Њ–≥—Г—В –Њ—Б—В–∞–≤–ї—П—В—М –њ–Њ—Б–ї–µ —Б–µ–±—П (–Њ—Б–Њ–±–µ–љ–љ–Њ –њ—А–Є –≥–Є–њ–Њ–≥–ї–Є–Ї–µ–Љ–Є—З–µ—Б–Ї–Њ–є –Ї–Њ–Љ–µ) —А–µ–Ј–Є–і—Г–∞–ї—М–љ—Л–є –і–µ—Д–µ–Ї—В, –Ї–Њ—В–Њ—А—Л–є –Њ—В¬†—Н–њ–Є–Ј–Њ–і–∞ –Ї¬†—Н–њ–Є–Ј–Њ–і—Г –Љ–Њ–ґ–µ—В ¬Ђ–љ–∞–Ї–∞–њ–ї–Є–≤–∞—В—М—Б—П¬ї. –Ю—Б—В—А—Л–Љ –њ–Њ—А–∞–ґ–µ–љ–Є–µ–Љ —В–∞–Ї–ґ–µ —Б—З–Є—В–∞–µ—В—Б—П –Є–љ—Б—Г–ї—М—В –Є–ї–Є —В—А–∞–љ–Ј–Є—В–Њ—А–љ–∞—П –Є—И–µ–Љ–Є—З–µ—Б–Ї–∞—П –∞—В–∞–Ї–∞ (–Ґ–Ш–Р), —З–∞—Б—В–Њ—В–∞ –Ї–Њ—В–Њ—А—Л—Е —Г¬†–ї–Є—Ж —Б¬†–°–Ф —Б—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ –≤—Л—И–µ, —З–µ–Љ –≤¬†–њ–Њ–њ—Г–ї—П—Ж–Є–Є [2, 4, 5].
–Ъ¬†—Е—А–Њ–љ–Є—З–µ—Б–Ї–Є–Љ, –Ї–∞–Ї –њ—А–∞–≤–Є–ї–Њ, –њ—А–Њ–≥—А–µ–і–Є–µ–љ—В–љ—Л–Љ –њ–Њ—А–∞–ґ–µ–љ–Є—П–Љ –≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ –Њ—В–љ–Њ—Б—П—В –і–Є–∞–±–µ—В–Є—З–µ—Б–Ї—Г—О —Н–љ—Ж–µ—Д–∞–ї–Њ–њ–∞—В–Є—О (–Ф–≠) –Є¬†—В–∞–Ї –љ–∞–Ј—Л–≤–∞–µ–Љ—Л–µ –Ї–Њ–Љ–Њ—А–±–Є–і–љ—Л–µ —Б–Њ—Б—В–Њ—П–љ–Є—П (–њ—А–µ–ґ–і–µ –≤—Б–µ–≥–Њ –і–Є—Б—Ж–Є—А–Ї—Г–ї—П—В–Њ—А–љ—Г—О —Н–љ—Ж–µ—Д–∞–ї–Њ–њ–∞—В–Є—О (–Ф–≠–Я) –Є¬†–±–Њ–ї–µ–Ј–љ—М –Р–ї—М—Ж–≥–µ–є–Љ–µ—А–∞ (–С–Р)), –Ї–Њ—В–Њ—А—Л–µ –њ—А–µ–і—Б—В–∞–≤–ї—П—О—В —Б–Њ–±–Њ–є —Б–∞–Љ–Њ—Б—В–Њ—П—В–µ–ї—М–љ—Л–µ –љ–Њ–Ј–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ —Д–Њ—А–Љ—Л, –њ–∞—В–Њ–≥–µ–љ–µ—В–Є—З–µ—Б–Ї–Є —Б–≤—П–Ј–∞–љ–љ—Л–µ —Б¬†–°–Ф.
–Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –Ї–∞–Ї –њ–Њ–Ї–∞–Ј—Л–≤–∞–µ—В –Ї–ї–Є–љ–Є—З–µ—Б–Ї–∞—П –њ—А–∞–Ї—В–Є–Ї–∞, —Б—Г—Й–µ—Б—В–≤—Г—О—В –њ–µ—А–µ—Е–Њ–і–љ—Л–µ (—Б–Њ—З–µ—В–∞–љ–љ—Л–µ) —Д–Њ—А–Љ—Л. –Ю–љ–Є –Ј–∞–љ–Є–Љ–∞—О—В –њ—А–Њ–Љ–µ–ґ—Г—В–Њ—З–љ–Њ–µ –њ–Њ–ї–Њ–ґ–µ–љ–Є–µ –Љ–µ–ґ–і—Г –Ф–≠ –Є¬†–С–Р –Є–ї–Є –Ф–≠–Я.
–Ґ–µ—А–Љ–Є–љ ¬Ђ–і–Є–∞–±–µ—В–Є—З–µ—Б–Ї–∞—П —Н–љ—Ж–µ—Д–∞–ї–Њ–њ–∞—В–Є—П¬ї –њ—А–µ–і–ї–Њ–ґ–Є–ї R. de Jong –≤¬†1950 –≥., –Њ–њ–Є—Б–∞–≤—И–Є–є –Ї–Њ–≥–љ–Є—В–Є–≤–љ–Њ–µ —Б–љ–Є–ґ–µ–љ–Є–µ —Г¬†–Љ–Њ–ї–Њ–і–Њ–≥–Њ —З–µ–ї–Њ–≤–µ–Ї–∞ —Б¬†–°–Ф 1 —В–Є–њ–∞. –Я–Њ–і –Ф–≠ –њ–Њ–љ–Є–Љ–∞—О—В –Љ–µ–і–ї–µ–љ–љ–Њ –њ—А–Њ–≥—А–µ—Б—Б–Є—А—Г—О—Й–Є–µ –і–Є—Д—Д—Г–Ј–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –≤¬†–≥–Њ–ї–Њ–≤–љ–Њ–Љ –Љ–Њ–Ј–≥–µ, –љ–µ–њ–Њ—Б—А–µ–і—Б—В–≤–µ–љ–љ–Њ —Б–≤—П–Ј–∞–љ–љ—Л–µ —Б¬†–Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Є–Љ–Є –љ–∞—А—Г—И–µ–љ–Є—П–Љ–Є –Є¬†–њ—А–Њ—П–≤–ї—П—О—Й–Є–µ—Б—П –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л–Љ —Б–љ–Є–ґ–µ–љ–Є–µ–Љ, —Ж–µ—А–µ–±—А–∞–ї—М–љ–Њ–є –∞—В—А–Њ—Д–Є–µ–є –Є/–Є–ї–Є –і–Є—Д—Д—Г–Ј–љ—Л–Љ –њ–Њ—А–∞–ґ–µ–љ–Є–µ–Љ –±–µ–ї–Њ–≥–Њ –≤–µ—Й–µ—Б—В–≤–∞ –≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ [1, 5вАУ8].
–І–∞—Б—В–Њ—В–∞ –≤—Б—В—А–µ—З–∞–µ–Љ–Њ—Б—В–Є –Ф–≠ —Б—А–µ–і–Є –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–°–Ф –Ї–Њ–ї–µ–±–ї–µ—В—Б—П –Њ—В¬†5¬†–і–Њ¬†80%. –°—В–Њ–ї—М –Ј–љ–∞—З–Є—В–µ–ї—М–љ—Л–є —А–∞–Ј–±—А–Њ—Б –Њ–±—К—П—Б–љ—П–µ—В—Б—П –Њ—В—Б—Г—В—Б—В–≤–Є–µ–Љ –Њ–±—Й–µ–њ—А–Є–љ—П—В—Л—Е –Ї—А–Є—В–µ—А–Є–µ–≤ –і–Є–∞–≥–љ–Њ—Б—В–Є–Ї–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П. –Т¬†—З–Є—Б—В–Њ–Љ –≤–Є–і–µ –Ф–≠ –Ї–∞–Ї –і–Є—Б–Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Њ–µ —А–∞—Б—Б—В—А–Њ–є—Б—В–≤–Њ –љ–∞–±–ї—О–і–∞–µ—В—Б—П –њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–µ–љ–љ–Њ —Г¬†–±–Њ–ї—М–љ—Л—Е –°–Ф 1 —В–Є–њ–∞. –£¬†–і–∞–љ–љ–Њ–є –Ї–∞—В–µ–≥–Њ—А–Є–Є –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–µ –Њ–±—Г—Б–ї–Њ–≤–ї–µ–љ–Њ –њ—А–µ–ґ–і–µ –≤—Б–µ–≥–Њ –љ–µ—Н—Д—Д–µ–Ї—В–Є–≤–љ—Л–Љ –Ї–Њ–љ—В—А–Њ–ї–µ–Љ –≥–ї–Є–Ї–µ–Љ–Є–Є (–≥–Є–њ–µ—А–≥–ї–Є–Ї–µ–Љ–Є–µ–є –Є–ї–Є –≥–Є–њ–Њ–≥–ї–Є–Ї–µ–Љ–Є—З–µ—Б–Ї–Є–Љ–Є —Н–њ–Є–Ј–Њ–і–∞–Љ–Є). –£¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–°–Ф 2 —В–Є–њ–∞ –Ф–≠ –±–Њ–ї–µ–µ —З–µ–Љ –≤¬†80% —Б–ї—Г—З–∞–µ–≤ —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞–µ—В—Б—П —Ж–µ—А–µ–±—А–Њ–≤–∞—Б–Ї—Г–ї—П—А–љ–Њ–є –њ–∞—В–Њ–ї–Њ–≥–Є–µ–є, —З—В–Њ –Њ–±—К—П—Б–љ—П–µ—В—Б—П –±–Њ–ї–µ–µ —Б—В–∞—А—И–Є–Љ –≤–Њ–Ј—А–∞—Б—В–Њ–Љ –Є¬†—И–Є—А–Њ–Ї–Є–Љ —А–∞—Б–њ—А–Њ—Б—В—А–∞–љ–µ–љ–Є–µ–Љ —Б–Њ—Б—Г–і–Є—Б—В—Л—Е —Д–∞–Ї—В–Њ—А–Њ–≤ —А–Є—Б–Ї–∞ (–∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–є –≥–Є–њ–µ—А—В–µ–љ–Ј–Є–Є, –і–Є—Б–ї–Є–њ–Є–і–µ–Љ–Є–Є). –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –µ—Б–ї–Є –њ—А–Є –°–Ф 1 —В–Є–њ–∞ —А–µ—И–∞—О—Й—Г—О —А–Њ–ї—М –≤¬†—А–∞–Ј–≤–Є—В–Є–Є –Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Є—Е –љ–∞—А—Г—И–µ–љ–Є–є –Є¬†–і–Є—Б—Д—Г–љ–Ї—Ж–Є–Є –≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ –Є–≥—А–∞–µ—В –і–µ—Д–Є—Ж–Є—В –Є–љ—Б—Г–ї–Є–љ–∞, –њ—А–Є –°–Ф 2 —В–Є–њ–∞ вАУ –Є–љ—Б—Г–ї–Є–љ–Њ—А–µ–Ј–Є—Б—В–µ–љ—В–љ–Њ—Б—В—М [3, 6, 8].
–С–Њ–ї–µ–Ј–љ—М –Р–ї—М—Ж–≥–µ–є–Љ–µ—А–∞ вАУ –Ї–Њ–Љ–Њ—А–±–Є–і–љ–Њ–µ —Б–Њ—Б—В–Њ—П–љ–Є–µ –Є–ї–Є¬†–і–Є–∞–±–µ—В 3 —В–Є–њ–∞?
–°—Г—Й–µ—Б—В–≤—Г–µ—В –њ–∞—А–∞–ї–ї–µ–ї–Є–Ј–Љ –≤¬†—А–∞–Ј–≤–Є—В–Є–Є –°–Ф –Є¬†–С–Р, –Ї–Њ—В–Њ—А—Л–є –≤–Њ¬†–Љ–љ–Њ–≥–Њ–Љ –Њ–±—К—П—Б–љ—П–µ—В—Б—П –љ–∞—А—Г—И–µ–љ–Є–µ–Љ —Д—Г–љ–Ї—Ж–Є–Є –Є–љ—Б—Г–ї–Є–љ–∞. –Т¬†–Њ—В–ї–Є—З–Є–µ –Њ—В¬†–њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Є—Е —В–Ї–∞–љ–µ–є –≤¬†–≥–Њ–ї–Њ–≤–љ–Њ–Љ –Љ–Њ–Ј–≥–µ –Є–љ—Б—Г–ї–Є–љ –љ–µ¬†—Г—З–∞—Б—В–≤—Г–µ—В –≤¬†—Г—В–Є–ї–Є–Ј–∞—Ж–Є–Є –≥–ї—О–Ї–Њ–Ј—Л, –Њ–і–љ–∞–Ї–Њ —Г—З–∞—Б—В–≤—Г–µ—В –≤¬†—А–µ–≥—Г–ї—П—Ж–Є–Є –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л—Е –њ—А–Њ—Ж–µ—Б—Б–Њ–≤.
–†–µ—Ж–µ–њ—В–Њ—А—Л –Є–љ—Б—Г–ї–Є–љ–∞ –Њ–±–љ–∞—А—Г–ґ–µ–љ—Л –≤¬†–Ї–Њ—А–µ –≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ –Є¬†–≥–Є–њ–њ–Њ–Ї–∞–Љ–њ–µ. –Ш—Е¬†–∞–Ї—В–Є–≤–∞—Ж–Є—П –њ—А–Є–≤–Њ–і–Є—В –Ї¬†—Г—Б–Є–ї–µ–љ–Є—О –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л—Е –Є¬†–њ—А–µ–ґ–і–µ –≤—Б–µ–≥–Њ –Љ–љ–µ—Б—В–Є—З–µ—Б–Ї–Є—Е —Д—Г–љ–Ї—Ж–Є–є.
–°–љ–Є–ґ–µ–љ–Є–µ —Б¬†–≤–Њ–Ј—А–∞—Б—В–Њ–Љ –≤¬†–Љ–Њ–Ј–≥–µ –Ї–Њ–љ—Ж–µ–љ—В—А–∞—Ж–Є–Є –Є–љ—Б—Г–ї–Є–љ–∞ –Є¬†–Ї–Њ–ї–Є—З–µ—Б—В–≤–∞ –Є–љ—Б—Г–ї–Є–љ–Њ–≤—Л—Е —А–µ—Ж–µ–њ—В–Њ—А–Њ–≤ —З–∞—Б—В–Є—З–љ–Њ –Њ–±—К—П—Б–љ—П–µ—В –≤–Њ–Ј—А–∞—Б—В–љ—Г—О –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Г—О –і–Є—Б—Д—Г–љ–Ї—Ж–Є—О. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –њ—А–Є —В—П–ґ–µ–ї–Њ–є –С–Р –≤¬†–Љ–Њ–Ј–≥–µ –Њ—В–Љ–µ—З–µ–љ–Њ —Б–љ–Є–ґ–µ–љ–Є–µ —Г—А–Њ–≤–љ—П –Є–љ—Б—Г–ї–Є–љ–∞ –Є¬†–њ—А–Њ–Љ–µ–ґ—Г—В–Њ—З–љ—Л—Е –њ—А–Њ–і—Г–Ї—В–Њ–≤ –Є–љ—Б—Г–ї–Є–љ–Њ–≤–Њ–≥–Њ —Б–Є–≥–љ–∞–ї—М–љ–Њ–≥–Њ –њ—Г—В–Є. –≠—В–Њ–Љ—Г –Љ–Њ–ґ–µ—В —Б–њ–Њ—Б–Њ–±—Б—В–≤–Њ–≤–∞—В—М –љ–∞—А—Г—И–µ–љ–Є–µ —В—А–∞–љ—Б–њ–Њ—А—В–∞ –Є–љ—Б—Г–ї–Є–љ–∞ –≤¬†–Љ–Њ–Ј–≥ —З–µ—А–µ–Ј –≥–µ–Љ–∞—В–Њ—Н–љ—Ж–µ—Д–∞–ї–Є—З–µ—Б–Ї–Є–є –±–∞—А—М–µ—А –Є¬†—Б–љ–Є–ґ–µ–љ–Є–µ —Б–Є–љ—В–µ–Ј–∞ –Є–љ—Б—Г–ї–Є–љ–∞ –≤¬†–Љ–Њ–Ј–≥–µ. –Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–С–Р –њ—А–Є –љ–Њ—А–Љ–∞–ї—М–љ–Њ–Љ –Є–ї–Є –≤—Л—Б–Њ–Ї–Њ–Љ —Г—А–Њ–≤–љ–µ –њ–µ—А–Є—Д–µ—А–Є—З–µ—Б–Ї–Њ–≥–Њ –Є–љ—Б—Г–ї–Є–љ–∞ —Г—А–Њ–≤–µ–љ—М –Є–љ—Б—Г–ї–Є–љ–∞ –≤¬†—Ж–µ—А–µ–±—А–Њ—Б–њ–Є–љ–∞–ї—М–љ–Њ–є –ґ–Є–і–Ї–Њ—Б—В–Є –Њ–±—Л—З–љ–Њ —Б–љ–Є–ґ–µ–љ [9вАУ12]. –Я—А–Є –С–Р –Є¬†–°–Ф —В–∞–Ї–ґ–µ –≤—Л—П–≤–ї–µ–љ–Њ —Г–Љ–µ–љ—М—И–µ–љ–Є–µ —Г—А–Њ–≤–љ—П –Є–љ—Б—Г–ї–Є–Ј–Є–љ–∞ (–Є–љ—Б—Г–ї–Є–љ-–і–µ–≥—А–∞–і–Є—А—Г—О—Й–µ–≥–Њ —Д–µ—А–Љ–µ–љ—В–∞), —А–∞—Б—Й–µ–њ–ї—П—О—Й–µ–≥–Њ –Ї–∞–Ї –Є–љ—Б—Г–ї–Є–љ, —В–∞–Ї –Є¬†–∞–Љ–Є–ї–Њ–Є–і–љ—Л–є –±–µ–ї–Њ–Ї, —З—В–Њ –љ–∞—А—Г—И–∞–µ—В –Ї–ї–Є—А–µ–љ—Б –∞–Љ–Є–ї–Њ–Є–і–љ–Њ–≥–Њ –±–µ–ї–Ї–∞ –Є–Ј¬†–≤–µ—Й–µ—Б—В–≤–∞ –≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞.
–Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –Є–љ—Б—Г–ї–Є–љ –Є–љ–∞–Ї—В–Є–≤–Є—А—Г–µ—В –Ї–Є–љ–∞–Ј—Г-3 –≥–ї–Є–Ї–Њ–≥–µ–љ—Б–Є–љ—В–µ—В–∞–Ј—Л, –њ—А–µ–њ—П—В—Б—В–≤—Г—П —Д–Њ—Б—Д–Њ—А–Є–ї–Є—А–Њ–≤–∞–љ–Є—О —В–∞—Г-–њ—А–Њ—В–µ–Є–љ–∞ –Є¬†—В–µ–Љ —Б–∞–Љ—Л–Љ —В–Њ—А–Љ–Њ–Ј—П –љ–∞–Ї–Њ–њ–ї–µ–љ–Є–µ –Є¬†–∞–≥—А–µ–≥–∞—Ж–Є—О —Г–Ї–∞–Ј–∞–љ–љ–Њ–≥–Њ –±–µ–ї–Ї–∞.
–Ш–љ—Б—Г–ї–Є–љ–Њ—А–µ–Ј–Є—Б—В–µ–љ—В–љ–Њ—Б—В—М —Б–≤—П–Ј–∞–љ–∞ —Б¬†–љ–∞—А—Г—И–µ–љ–Є–µ–Љ –Њ–±–Љ–µ–љ–∞ –∞–Љ–Є–ї–Њ–Є–і–∞, –∞—В—А–Њ—Д–Є–µ–є –≥–Є–њ–њ–Њ–Ї–∞–Љ–њ–∞, –љ–∞—А—Г—И–µ–љ–Є–µ–Љ –њ–∞–Љ—П—В–Є –Є¬†–њ–Њ–≤—Л—И–µ–љ–Є–µ–Љ —А–Є—Б–Ї–∞ —А–∞–Ј–≤–Є—В–Є—П –С–Р –≤¬†–њ–Њ–ї—В–Њ—А–∞ вАУ –і–≤–∞ —А–∞–Ј–∞. –Ю¬†—А–Њ–ї–Є –Є–љ—Б—Г–ї–Є–љ–∞ –≤¬†—А–µ–≥—Г–ї–Є—А–Њ–≤–∞–љ–Є–Є –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л—Е –њ—А–Њ—Ж–µ—Б—Б–Њ–≤ —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г–µ—В —Г–ї—Г—З—И–µ–љ–Є–µ –њ—А–Њ—Ж–µ—Б—Б–Њ–≤ –њ–∞–Љ—П—В–Є –њ—А–Є –µ–≥–Њ –≤–љ—Г—В—А–Є–≤–µ–љ–љ–Њ–Љ –Є–ї–Є –Є–љ—В—А–∞–љ–∞–Ј–∞–ї—М–љ–Њ–Љ –≤–≤–µ–і–µ–љ–Є–Є [1, 7, 13].
–Ю–±—Й–Є–Љ–Є —Д–∞–Ї—В–Њ—А–∞–Љ–Є –њ–∞—В–Њ–≥–µ–љ–µ–Ј–∞ –°–Ф –Є¬†–С–Р —П–≤–ї—П—О—В—Б—П –Њ–Ї–Є—Б–ї–Є—В–µ–ї—М–љ—Л–є —Б—В—А–µ—Б—Б –Є¬†–њ–Њ–≤—Л—И–µ–љ–љ–Њ–µ –Њ–±—А–∞–Ј–Њ–≤–∞–љ–Є–µ –Ї–Њ–љ–µ—З–љ—Л—Е –њ—А–Њ–і—Г–Ї—В–Њ–≤ –≥–ї–Є–Ї–Є—А–Њ–≤–∞–љ–Є—П. –Э–∞–±–ї—О–і–∞–µ—В—Б—П —В–∞–Ї–ґ–µ —Б—Е–Њ–і—Б—В–≤–Њ –≤¬†–і–Є–Ј—А–µ–≥—Г–ї—П—Ж–Є–Є –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ–∞ –≥–ї—О–Ї–Њ–Ј—Л, –љ–∞—А—Г—И–µ–љ–Є–Є –Њ–±–Љ–µ–љ–∞ –∞–њ–Њ–ї–Є–њ–Њ–њ—А–Њ—В–µ–Є–љ–∞ –Х, –Љ–Є—В–Њ—Е–Њ–љ–і—А–Є–∞–ї—М–љ–Њ–є –і–Є—Б—Д—Г–љ–Ї—Ж–Є–Є, –љ–∞—А—Г—И–µ–љ–Є–Є –≥–Њ–Љ–µ–Њ—Б—В–∞–Ј–∞ –Ї–∞–ї—М—Ж–Є—П. –Э–∞¬†—Н–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–∞–ї—М–љ–Њ–є –Љ–Њ–і–µ–ї–Є –њ–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ –°–Ф —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞–µ—В—Б—П –њ–Њ—А–∞–ґ–µ–љ–Є–µ–Љ –љ–µ–є—А–Њ–љ–Њ–≤ –≥–Є–њ–њ–Њ–Ї–∞–Љ–њ–∞ –Є¬†–Љ–Є–љ–і–∞–ї–Є–љ—Л вАУ –Ј–Њ–љ, –Ї–Њ—В–Њ—А—Л–µ —Б—В—А–∞–і–∞—О—В –љ–∞¬†—А–∞–љ–љ–µ–Љ —Н—В–∞–њ–µ —А–∞–Ј–≤–Є—В–Є—П –С–Р. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –њ–∞—Ж–Є–µ–љ—В—Л —Б¬†–С–Р –њ—А–µ–і—А–∞—Б–њ–Њ–ї–Њ–ґ–µ–љ—Л –Ї¬†—А–∞–Ј–≤–Є—В–Є—О –°–Ф 2 —В–Є–њ–∞ –Є¬†—З–∞—Й–µ –Є–Љ–µ—О—В –≥–Є–њ–µ—А–Є–љ—Б—Г–ї–Є–љ–µ–Љ–Є—О –Є¬†–≥–Є–њ–µ—А–≥–ї–Є–Ї–µ–Љ–Є—О –њ–Њ¬†—Б—А–∞–≤–љ–µ–љ–Є—О —Б–Њ¬†–Ј–і–Њ—А–Њ–≤—Л–Љ–Є –ї–Є—Ж–∞–Љ–Є —В–Њ–≥–Њ –ґ–µ –≤–Њ–Ј—А–∞—Б—В–∞ [8].
–°–Њ–Њ—В–љ–Њ—И–µ–љ–Є–µ —А–Њ–ї–Є –Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Є—Е –љ–∞—А—Г—И–µ–љ–Є–є –Є¬†—Б–Њ—Б—Г–і–Є—Б—В—Л—Е —Д–∞–Ї—В–Њ—А–Њ–≤ —А–Є—Б–Ї–∞ –≤¬†–њ–Њ—А–∞–ґ–µ–љ–Є–Є –≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ –њ—А–Є —Б–∞—Е–∞—А–љ–Њ–Љ –і–Є–∞–±–µ—В–µ
–°–ї–Њ–ґ–љ–Њ —Б–Њ–њ–Њ—Б—В–∞–≤–Є—В—М –Ј–љ–∞—З–Є–Љ–Њ—Б—В—М —Б–Њ–±—Б—В–≤–µ–љ–љ–Њ –Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Є—Е —А–∞—Б—Б—В—А–Њ–є—Б—В–≤ –Є¬†—Б–Њ—Б—Г–і–Є—Б—В—Л—Е —Д–∞–Ї—В–Њ—А–Њ–≤ —А–Є—Б–Ї–∞, —З–∞—Б—В–Њ –љ–∞–±–ї—О–і–∞—О—Й–Є—Е—Б—П —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–°–Ф 2 —В–Є–њ–∞, –≤¬†—А–∞–Ј–≤–Є—В–Є–Є –∞—В—А–Њ—Д–Є–Є –≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ –Є–ї–Є –ї–µ–є–Ї–Њ—Н–љ—Ж–µ—Д–∞–ї–Њ–њ–∞—В–Є–Є, –≤—Л—П–≤–ї—П–µ–Љ—Л—Е –њ—А–Є –љ–µ–є—А–Њ–≤–Є–Ј—Г–∞–ї–Є–Ј–∞—Ж–Є–Є, –Є¬†—Б–≤—П–Ј–∞–љ–љ—Л—Е —Б¬†–љ–Є–Љ–Є –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л—Е –љ–∞—А—Г—И–µ–љ–Є–є.
–Ю—В–њ—А–∞–≤–љ–Њ–є —В–Њ—З–Ї–Њ–є –Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Њ–≥–Њ –њ—Г—В–Є –њ–Њ—А–∞–ґ–µ–љ–Є—П –≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞, –≤–µ—А–Њ—П—В–љ–Њ, —П–≤–ї—П–µ—В—Б—П –≥–Є–њ–µ—А–≥–ї–Є–Ї–µ–Љ–Є—П, –Њ–і–љ–∞–Ї–Њ –Ї–Њ–љ–Ї—А–µ—В–љ—Л–µ –Љ–µ—Е–∞–љ–Є–Ј–Љ—Л –Љ–Њ–≥—Г—В –±—Л—В—М —Б–≤—П–Ј–∞–љ—Л —Б¬†–љ–∞–Ї–Њ–њ–ї–µ–љ–Є–µ–Љ –Ї–Њ–љ–µ—З–љ—Л—Е –њ—А–Њ–і—Г–Ї—В–Њ–≤ –≥–ї–Є–Ї–Є—А–Њ–≤–∞–љ–Є—П, –∞–Ї—В–Є–≤–љ—Л—Е —Д–Њ—А–Љ –Ї–Є—Б–ї–Њ—А–Њ–і–∞ –Є¬†—Б–≤–Њ–±–Њ–і–љ—Л—Е —А–∞–і–Є–Ї–∞–ї–Њ–≤ (–Њ–Ї–Є—Б–ї–Є—В–µ–ї—М–љ—Л–є —Б—В—А–µ—Б—Б), –і–Є—Б—Д—Г–љ–Ї—Ж–Є–µ–є –Є–љ—Б—Г–ї–Є–љ–Њ–≤–Њ–≥–Њ —Б–Є–≥–љ–∞–ї—М–љ–Њ–≥–Њ –њ—Г—В–Є. –Ґ–Њ–Ї—Б–Є—З–µ—Б–Ї–Њ–µ –≤–Њ–Ј–і–µ–є—Б—В–≤–Є–µ –Є–Ј–±—Л—В–Ї–∞ –≥–ї—О–Ї–Њ–Ј—Л –Љ–Њ–ґ–µ—В –±—Л—В—М —Б–≤—П–Ј–∞–љ–Њ —Б¬†–∞–Ї—В–Є–≤–∞—Ж–Є–µ–є –њ–Њ–ї–Є–Њ–ї–Њ–≤–Њ–≥–Њ –Є¬†–≥–µ–Ї—Б–Њ–Ј–∞–Љ–Є–љ–Њ–≤–Њ–≥–Њ –њ—Г—В–µ–є. –Я—А–Є —Г–ї—Г—З—И–µ–љ–Є–Є –Ї–Њ–љ—В—А–Њ–ї—П –°–Ф –Ї–Њ–≥–љ–Є—В–Є–≤–љ–∞—П –і–Є—Б—Д—Г–љ–Ї—Ж–Є—П, –Њ—В—А–∞–ґ–∞—О—Й–∞—П –њ–Њ—А–∞–ґ–µ–љ–Є–µ –Љ–Њ–Ј–≥–∞, —З–∞—Б—В–Є—З–љ–Њ –Њ–±—А–∞—В–Є–Љ–∞.
–†–Њ–ї—М –≥–Є–њ–Њ–≥–ї–Є–Ї–µ–Љ–Є–Є –≤¬†—А–∞–Ј–≤–Є—В–Є–Є –∞—В—А–Њ—Д–Є–Є –≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ –Є–ї–Є –ї–µ–є–Ї–Њ—Н–љ—Ж–µ—Д–∞–ї–Њ–њ–∞—В–Є–Є –њ–Њ–і—В–≤–µ—А–ґ–і–µ–љ–∞ –љ–µ¬†–≤–Њ –≤—Б–µ—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П—Е, —З—В–Њ, –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ, –Њ—В—А–∞–ґ–∞–µ—В –њ–Њ–Ј–Є—В–Є–≤–љ–Њ–µ –≤–ї–Є—П–љ–Є–µ –Є–љ—Б—Г–ї–Є–љ–∞ –љ–∞¬†–Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л–µ —Д—Г–љ–Ї—Ж–Є–Є [11, 14, 15].
–Р—В—А–Њ—Д–Є—П –Љ–Њ–Ј–≥–∞ –Є¬†–і–Є—Д—Д—Г–Ј–љ–Њ–µ –њ–Њ—А–∞–ґ–µ–љ–Є–µ –±–µ–ї–Њ–≥–Њ –≤–µ—Й–µ—Б—В–≤–∞, –≤—Л—П–≤–ї—П–µ–Љ—Л–µ –њ—А–Є –Љ–∞–≥–љ–Є—В–љ–Њ-—А–µ–Ј–Њ–љ–∞–љ—Б–љ–Њ–є —В–Њ–Љ–Њ–≥—А–∞—Д–Є–Є, –∞—Б—Б–Њ—Ж–Є–Є—А–Њ–≤–∞–љ—Л —Б¬†–љ–∞—А—Г—И–µ–љ–Є–µ–Љ –Љ–Є–µ–ї–Є–љ–Є–Ј–∞—Ж–Є–Є –љ–µ—А–≤–љ—Л—Е –≤–Њ–ї–Њ–Ї–Њ–љ. –†–µ—Ж–µ–њ—В–Њ—А—Л –Ї–Њ–љ–µ—З–љ—Л—Е –њ—А–Њ–і—Г–Ї—В–Њ–≤ –≥–ї–Є–Ї–Є—А–Њ–≤–∞–љ–Є—П —Н–Ї—Б–њ—А–µ—Б—Б–Є—А–Њ–≤–∞–љ—Л –Ї–∞–Ї –≤¬†—Б–µ—А–Њ–Љ –≤–µ—Й–µ—Б—В–≤–µ (–≥–Є–њ–њ–Њ–Ї–∞–Љ–њ–µ –Є¬†–Ї–Њ—А–µ –≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞), —В–∞–Ї –Є¬†–≤ –±–µ–ї–Њ–Љ, –Њ—Б–Њ–±–µ–љ–љ–Њ –≤¬†–Љ–Њ–Ј–Њ–ї–Є—Б—В–Њ–Љ —В–µ–ї–µ –Є¬†–≤–љ—Г—В—А–µ–љ–љ–µ–є –Ї–∞–њ—Б—Г–ї–µ. –Я–Њ–≤—Л—И–µ–љ–љ–Њ–µ –Ї–Њ–ї–Є—З–µ—Б—В–≤–Њ —Н—В–Є—Е —А–µ—Ж–µ–њ—В–Њ—А–Њ–≤ –Њ–±—К—П—Б–љ—П–µ—В—Б—П —Г—Б—В—А–∞–љ–µ–љ–Є–µ–Љ —В–Њ—А–Љ–Њ–ґ–µ–љ–Є—П –њ–Њ–і –≤–ї–Є—П–љ–Є–µ–Љ –љ—Г–Ї–ї–µ–∞—А–љ–Њ–≥–Њ (—П–і–µ—А–љ–Њ–≥–Њ) —Д–∞–Ї—В–Њ—А–∞ ќЇB, —З—В–Њ —Б–≤—П–Ј–∞–љ–Њ —Б¬†–і–Є—Б—Д—Г–љ–Ї—Ж–Є–µ–є –Є–љ—Б—Г–ї–Є–љ–Њ–≤–Њ–≥–Њ —Б–Є–≥–љ–∞–ї—М–љ–Њ–≥–Њ –њ—Г—В–Є. –≠—В–Њ—В –ґ–µ —Д–∞–Ї—В–Њ—А –≤–ї–Є—П–µ—В –љ–∞¬†–њ—А–Њ–і—Г–Ї—Ж–Є—О —Д–∞–Ї—В–Њ—А–∞ –љ–µ–Ї—А–Њ–Ј–∞ –Њ–њ—Г—Е–Њ–ї–Є –∞–ї—М—Д–∞, –Ї–Њ—В–Њ—А—Л–є —В–∞–Ї–ґ–µ —Г—З–∞—Б—В–≤—Г–µ—В –≤¬†—А–∞–Ј–≤–Є—В–Є–Є –і–Є–∞–±–µ—В–Є—З–µ—Б–Ї–Њ–є –ї–µ–є–Ї–Њ—Н–љ—Ж–µ—Д–∞–ї–Њ–њ–∞—В–Є–Є. –Ю–і–љ–∞–Ї–Њ –ї–µ–є–Ї–Њ—Н–љ—Ж–µ—Д–∞–ї–Њ–њ–∞—В–Є—П –њ—А–Є –°–Ф –Љ–Њ–ґ–µ—В –±—Л—В—М –≤—Л–Ј–≤–∞–љ–∞ –Є¬†–∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–є –≥–Є–њ–µ—А—В–µ–љ–Ј–Є–µ–є. –Ґ–µ–Љ –љ–µ¬†–Љ–µ–љ–µ–µ —А–µ–Ј—Г–ї—М—В–∞—В—Л –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –њ–Њ–Ї–∞–Ј—Л–≤–∞—О—В, —З—В–Њ –Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Є–µ –љ–∞—А—Г—И–µ–љ–Є—П, —Б–≤—П–Ј–∞–љ–љ—Л–µ —Б¬†–°–Ф, —В–µ—Б–љ–µ–µ –Ї–Њ—А—А–µ–ї–Є—А—Г—О—В —Б¬†—А–∞–Ј–≤–Є—В–Є–µ–Љ —Ж–µ—А–µ–±—А–∞–ї—М–љ–Њ–є –∞—В—А–Њ—Д–Є–Є –Є¬†–Є–Ј–Љ–µ–љ–µ–љ–Є–µ–Љ –±–µ–ї–Њ–≥–Њ –≤–µ—Й–µ—Б—В–≤–∞, —В–Њ–≥–і–∞ –Ї–∞–Ї –∞—А—В–µ—А–Є–∞–ї—М–љ–∞—П –≥–Є–њ–µ—А—В–µ–љ–Ј–Є—П –∞—Б—Б–Њ—Ж–Є–Є—А–Њ–≤–∞–љ–∞ –ї–Є—И—М —Б¬†–ї–µ–≥–Ї–Є–Љ –Є–Ј–Љ–µ–љ–µ–љ–Є–µ–Љ —З–Є—Б–ї–µ–љ–љ–Њ—Б—В–Є –љ–µ–є—А–Њ–љ–Њ–≤ –≥–Є–њ–њ–Њ–Ї–∞–Љ–њ–∞ –Є¬†—Г—В—А–∞—В–Њ–є —Б–Є–љ–∞–њ—Б–Њ–≤. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л–µ –љ–∞—А—Г—И–µ–љ–Є—П –њ—А–Є –°–Ф –≤—Л—А–∞–ґ–µ–љ—Л –≤¬†–±–Њ–ї—М—И–µ–є —Б—В–µ–њ–µ–љ–Є –Є¬†–љ–∞—Б—В—Г–њ–∞—О—В –±—Л—Б—В—А–µ–µ, —З–µ–Љ –њ—А–Є –љ–∞–ї–Є—З–Є–Є —В–Њ–ї—М–Ї–Њ –∞—А—В–µ—А–Є–∞–ї—М–љ–Њ–є –≥–Є–њ–µ—А—В–µ–љ–Ј–Є–Є. –Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, –Ї–∞–Ї –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є–µ, —В–∞–Ї –Є¬†—Н–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–∞–ї—М–љ—Л–µ –і–∞–љ–љ—Л–µ —Г–Ї–∞–Ј—Л–≤–∞—О—В –љ–∞¬†—Б–Є–љ–µ—А–≥–Є–Ј–Љ –Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Є—Е –љ–∞—А—Г—И–µ–љ–Є–є –Є¬†–≥–Є–њ–µ—А—В–µ–љ–Ј–Є–Є –≤¬†–њ–Њ—А–∞–ґ–µ–љ–Є–Є –Љ–Њ–Ј–≥–∞ [6, 8, 11, 13, 16].
–†–Њ–ї—М –≥–Є–њ–µ—А–ї–Є–њ–Є–і–µ–Љ–Є–Є –≤¬†—А–∞–Ј–≤–Є—В–Є–Є –Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Њ–≥–Њ –њ–Њ—А–∞–ґ–µ–љ–Є—П –Љ–Њ–Ј–≥–∞ –љ–µ¬†–Њ–њ—А–µ–і–µ–ї–µ–љ–∞. –Ш–Ј–≤–µ—Б—В–љ–Њ, —З—В–Њ –њ–Њ–≤—Л—И–µ–љ–љ—Л–є —Г—А–Њ–≤–µ–љ—М —В—А–Є–≥–ї–Є—Ж–µ—А–Є–і–Њ–≤ —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г–µ—В –њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є—О –і–Є–∞–±–µ—В–Є—З–µ—Б–Ї–Њ–є –љ–µ–≤—А–Њ–њ–∞—В–Є–Є. –Э–∞—А—Г—И–µ–љ–Є–µ –Љ–µ—В–∞–±–Њ–ї–Є–Ј–Љ–∞ –ї–Є–њ–Є–і–Њ–≤ –Љ–Њ–ґ–µ—В –≤–љ–Њ—Б–Є—В—М —Б—Г—Й–µ—Б—В–≤–µ–љ–љ—Л–є –≤–Ї–ї–∞–і –≤¬†–Ј–≤–µ–љ—М—П –њ–∞—В–Њ–≥–µ–љ–µ–Ј–∞, —Б–≤—П–Ј–∞–љ–љ—Л–µ —Б¬†–Ї–Њ–љ–µ—З–љ—Л–Љ–Є –њ—А–Њ–і—Г–Ї—В–∞–Љ–Є –≥–ї–Є–Ї–Є—А–Њ–≤–∞–љ–Є—П. –Ю–Ї—Б–Є–і–∞—Ж–Є—П –ї–Є–њ–Њ–њ—А–Њ—В–µ–Є–љ–Њ–≤ –љ–Є–Ј–Ї–Њ–є –њ–ї–Њ—В–љ–Њ—Б—В–Є –Љ–Њ–ґ–µ—В –Є–≥—А–∞—В—М —А–µ—И–∞—О—Й—Г—О —А–Њ–ї—М –≤¬†—А–∞–Ј–≤–Є—В–Є–Є –∞—В–µ—А–Њ—Б–Ї–ї–µ—А–Њ–Ј–∞, –Њ–Ї–Є—Б–ї–Є—В–µ–ї—М–љ–Њ–≥–Њ —Б—В—А–µ—Б—Б–∞ –Є¬†–≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л—Е –њ—А–Њ—Ж–µ—Б—Б–Њ–≤ [15, 16].
–†–Њ–ї—М —Б–Њ—Б—Г–і–Є—Б—В–Њ–≥–Њ —Д–∞–Ї—В–Њ—А–∞ –≤¬†—А–∞–Ј–≤–Є—В–Є–Є –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л—Е –љ–∞—А—Г—И–µ–љ–Є–є –њ—А–Є –°–Ф –Љ–Њ–ґ–µ—В –њ—А–µ—Г–≤–µ–ї–Є—З–Є–≤–∞—В—М—Б—П –≤¬†—Б–Є–ї—Г –±–Њ–ї–µ–µ –њ—А–Њ—Б—В–Њ–≥–Њ –≤—Л—П–≤–ї–µ–љ–Є—П —Б–Њ—Б—Г–і–Є—Б—В—Л—Е –Є–Ј–Љ–µ–љ–µ–љ–Є–є –Љ–Њ–Ј–≥–∞ –њ—А–Є –≤–Є–Ј—Г–∞–ї–Є–Ј–∞—Ж–Є–Є. –£¬†–±–Њ–ї—М–љ—Л—Е –°–Ф –і–Є—Д—Д—Г–Ј–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –±–µ–ї–Њ–≥–Њ –≤–µ—Й–µ—Б—В–≤–∞, –љ–µ–Љ—Л–µ –Є–љ—Д–∞—А–Ї—В—Л –Є¬†—Ж–µ—А–µ–±—А–∞–ї—М–љ–∞—П –∞—В—А–Њ—Д–Є—П –Љ–Њ–≥—Г—В –Њ—В—А–∞–ґ–∞—В—М –≤–Њ–Ј—А–∞—Б—В–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П, –Ї–Њ—В–Њ—А—Л–µ –љ–µ¬†–Њ–±—П–Ј–∞—В–µ–ї—М–љ–Њ —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞—О—В—Б—П –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л–Љ —Б–љ–Є–ґ–µ–љ–Є–µ–Љ. –Э–∞¬†—Н–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–∞–ї—М–љ—Л—Е –Љ–Њ–і–µ–ї—П—Е –Ф–≠ –љ–µ¬†–≤—Л—П–≤–ї–µ–љ–Њ —Б–љ–Є–ґ–µ–љ–Є—П –њ–µ—А—Д—Г–Ј–Є–Є –Љ–Њ–Ј–≥–∞. –Ш–Ј–Љ–µ–љ–µ–љ–Є–є –њ–µ—А—Д—Г–Ј–Є–Є –љ–µ¬†—Г—Б—В–∞–љ–Њ–≤–ї–µ–љ–Њ –Є¬†–≤ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Є—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П—Е [7, 9, 17].
–Т¬†—В–Њ –ґ–µ –≤—А–µ–Љ—П —Г¬†–±–Њ–ї—М–љ—Л—Е –°–Ф 2 —В–Є–њ–∞, –њ—А–Є –Ї–Њ—В–Њ—А–Њ–Љ —З–∞—Й–µ –Њ—В–Љ–µ—З–∞—О—В—Б—П –∞—В—А–Њ—Д–Є—П –њ–Њ–і–Ї–Њ—А–Ї–Њ–≤—Л—Е —Б—В—А—Г–Ї—В—Г—А –Є¬†–њ–∞—В–Њ–ї–Њ–≥–Є—П –±–µ–ї–Њ–≥–Њ –≤–µ—Й–µ—Б—В–≤–∞, –Ї–∞–Ї –њ—А–∞–≤–Є–ї–Њ, –Њ—В–Љ–µ—З–∞–µ—В—Б—П –і–Є—Б—А–µ–≥—Г–ї—П—В–Њ—А–љ—Л–є (–њ–Њ–і–Ї–Њ—А–Ї–Њ–≤–Њ-–ї–Њ–±–љ—Л–є) –њ—А–Њ—Д–Є–ї—М –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л—Е –љ–∞—А—Г—И–µ–љ–Є–є. –Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, –Ї–Њ—А—А–µ–ї—П—Ж–Є—П –Љ–µ–ґ–і—Г —Е–∞—А–∞–Ї—В–µ—А–Њ–Љ –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л—Е –љ–∞—А—Г—И–µ–љ–Є–є –Є¬†–љ–µ–є—А–Њ–≤–Є–Ј—Г–∞–ї–Є–Ј–∞—Ж–Є–Њ–љ–љ—Л–Љ–Є –Є–Ј–Љ–µ–љ–µ–љ–Є—П–Љ–Є –Љ–Њ–ґ–µ—В –Њ—В—А–∞–ґ–∞—В—М –≤–Ї–ї–∞–і —Б–Њ—Б—Г–і–Є—Б—В—Л—Е, –і–Є—Б–Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Є—Е –Є¬†–і–µ–≥–µ–љ–µ—А–∞—В–Є–≤–љ—Л—Е –Є–Ј–Љ–µ–љ–µ–љ–Є–є –≤¬†—Д–Њ—А–Љ–Є—А–Њ–≤–∞–љ–Є–µ –Ї–ї–Є–љ–Є—З–µ—Б–Ї–Њ–є –Ї–∞—А—В–Є–љ—Л [11, 15, 17].
–Ъ–Њ–≥–љ–Є—В–Є–≤–љ–∞—П –і–Є—Б—Д—Г–љ–Ї—Ж–Є—П¬†вАУ –Њ—Б–љ–Њ–≤–љ–Њ–µ –њ—А–Њ—П–≤–ї–µ–љ–Є–µ –і–Є–∞–±–µ—В–Є—З–µ—Б–Ї–Њ–є —Н–љ—Ж–µ—Д–∞–ї–Њ–њ–∞—В–Є–Є
–Ю—Б–љ–Њ–≤–љ—Л–Љ –њ—А–Њ—П–≤–ї–µ–љ–Є–µ–Љ –љ–∞—А—Г—И–µ–љ–Є–є –≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ –њ—А–Є –°–Ф —Б–ї—Г–ґ–∞—В –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л–µ –Є¬†–∞—Д—Д–µ–Ї—В–Є–≤–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П, —З—В–Њ –Њ—В—А–∞–ґ–∞–µ—В –њ—А–µ–Є–Љ—Г—Й–µ—Б—В–≤–µ–љ–љ—Г—О –і–Є—Б—Д—Г–љ–Ї—Ж–Є—О —Д—А–Њ–љ—В–Њ—Б—В—А–Є–∞—А–љ—Л—Е –Ї—А—Г–≥–Њ–≤, —А–µ–≥—Г–ї–Є—А—Г—О—Й–Є—Е —Б–Њ–Њ—В–≤–µ—В—Б—В–≤—Г—О—Й–Є–µ –љ–µ–є—А–Њ–њ—Б–Є—Е–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –њ—А–Њ—Ж–µ—Б—Б—Л. –Я—А–Є –°–Ф –љ–∞—А—Г—И–∞—О—В—Б—П –њ–∞–Љ—П—В—М, –≤–љ–Є–Љ–∞–љ–Є–µ, –Ј—А–Є—В–µ–ї—М–љ–Њ-–њ—А–Њ—Б—В—А–∞–љ—Б—В–≤–µ–љ–љ—Л–µ –Є¬†—А–µ–≥—Г–ї—П—В–Њ—А–љ—Л–µ —Д—Г–љ–Ї—Ж–Є–Є.
–Ю¬†–њ—А–Њ–≥—А–µ—Б—Б–Є—А–Њ–≤–∞–љ–Є–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г–µ—В –њ—А–µ–ґ–і–µ –≤—Б–µ–≥–Њ —Б–љ–Є–ґ–µ–љ–Є–µ –њ–∞–Љ—П—В–Є (–Њ—Б–Њ–±–µ–љ–љ–Њ –њ—А–Є –њ—А–Њ–≤–µ–і–µ–љ–Є–Є —В–µ—Б—В–Њ–≤ –љ–∞¬†–Њ—В—Б—А–Њ—З–µ–љ–љ–Њ–µ –≤–Њ—Б–њ—А–Њ–Є–Ј–≤–µ–і–µ–љ–Є–µ). –Т¬†–њ–Њ–≤—Б–µ–і–љ–µ–≤–љ–Њ–є –ґ–Є–Ј–љ–Є –≤¬†–±–Њ–ї—М—И–µ–є —Б—В–µ–њ–µ–љ–Є –њ—А–Њ—П–≤–ї—П–µ—В—Б—П –і–Є—Б—А–µ–≥—Г–ї—П—В–Њ—А–љ—Л–є –і–µ—Д–Є—Ж–Є—В, –њ—А–µ–і–Њ–њ—А–µ–і–µ–ї—П—О—Й–Є–є –љ–µ—Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В—М –њ–∞—Ж–Є–µ–љ—В–Њ–≤ –Њ–±—А–∞–±–∞—В—Л–≤–∞—В—М –љ–µ—Б—В—А—Г–Ї—В—Г—А–Є—А–Њ–≤–∞–љ–љ—Г—О –Є–љ—Д–Њ—А–Љ–∞—Ж–Є—О, –Њ—Б–Њ–±–µ–љ–љ–Њ –≤¬†—Б–Є—В—Г–∞—Ж–Є—П—Е, —В—А–µ–±—Г—О—Й–Є—Е –љ–µ–Љ–µ–і–ї–µ–љ–љ—Л—Е —А–µ—И–µ–љ–Є–є.
–С–Њ–ї—М—И–Є–љ—Б—В–≤–Њ –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–°–Ф 2¬†—В–Є–њ–∞ –љ–µ¬†—Б—В—А–∞–і–∞—О—В –і–µ–Љ–µ–љ—Ж–Є–µ–є, –Њ–і–љ–∞–Ї–Њ –і–µ–Љ–Њ–љ—Б—В—А–Є—А—Г—О—В –Њ–њ—А–µ–і–µ–ї–µ–љ–љ—Л–µ –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л–µ –Ј–∞—В—А—Г–і–љ–µ–љ–Є—П, –Ї–Њ—В–Њ—А—Л–µ –Љ–Њ–ґ–љ–Њ —Е–∞—А–∞–Ї—В–µ—А–Є–Ј–Њ–≤–∞—В—М –Ї–∞–Ї —Г–Љ–µ—А–µ–љ–љ—Л–µ –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л–µ —А–∞—Б—Б—В—А–Њ–є—Б—В–≤–∞. –Т¬†—В–Њ –ґ–µ –≤—А–µ–Љ—П —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†—Г–Љ–µ—А–µ–љ–љ—Л–Љ–Є –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л–Љ–Є —А–∞—Б—Б—В—А–Њ–є—Б—В–≤–∞–Љ–Є –њ–Њ–≤—Л—И–µ–љ —А–Є—Б–Ї –Ї–Њ–љ–≤–µ—А—Б–Є–Є –Є—Е –≤¬†–і–µ–Љ–µ–љ—Ж–Є—О [1, 8, 9].
–Я—А–Є –°–Ф 1 —В–Є–њ–∞, –і–µ–±—О—В–Є—А—Г—О—Й–µ–Љ –≤¬†–і–µ—В—Б–Ї–Њ–Љ –≤–Њ–Ј—А–∞—Б—В–µ, –Њ—В–Љ–µ—З–∞—О—В—Б—П –Ј–∞–Љ–µ–і–ї–µ–љ–Є–µ –Є–љ—В–µ–ї–ї–µ–Ї—В—Г–∞–ї—М–љ–Њ–≥–Њ —А–∞–Ј–≤–Є—В–Є—П –Є¬†—В—А—Г–і–љ–Њ—Б—В–Є –Њ–±—Г—З–µ–љ–Є—П –≤¬†—И–Ї–Њ–ї–µ. –≠—В–Њ—В –і–µ—Д–µ–Ї—В –њ–µ—А—Б–Є—Б—В–Є—А—Г–µ—В –Є¬†–≤ –±–Њ–ї–µ–µ –Ј—А–µ–ї–Њ–Љ –≤–Њ–Ј—А–∞—Б—В–µ, –љ–µ—А–µ–і–Ї–Њ —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞—П—Б—М –і–∞–ї—М–љ–µ–є—И–Є–Љ –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л–Љ —Б–љ–Є–ґ–µ–љ–Є–µ–Љ –Є¬†–±–Њ–ї–µ–µ –≤—Л—А–∞–ґ–µ–љ–љ–Њ–є —Ж–µ—А–µ–±—А–∞–ї—М–љ–Њ–є –∞—В—А–Њ—Д–Є–µ–є. –Т–∞–ґ–љ—Г—О —А–Њ–ї—М –Є–≥—А–∞—О—В –њ–Њ–≤—В–Њ—А—П—О—Й–Є–µ—Б—П —Н–њ–Є–Ј–Њ–і—Л –≥–Є–њ–Њ–≥–ї–Є–Ї–µ–Љ–Є–Є, –Ї–Њ—В–Њ—А—Л–µ —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г—О—В —Г—Б—Г–≥—Г–±–ї–µ–љ–Є—О —Ж–µ—А–µ–±—А–∞–ї—М–љ–Њ–є –∞—В—А–Њ—Д–Є–Є.
–Я—А–Є –°–Ф 2 —В–Є–њ–∞ —Б–љ–Є–ґ–µ–љ–Є–µ –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л—Е —Д—Г–љ–Ї—Ж–Є–є –Љ–Њ–ґ–µ—В –њ—А–Њ–Є—Б—Е–Њ–і–Є—В—М –њ–∞—А–∞–ї–ї–µ–ї—М–љ–Њ —Б¬†–њ–µ—А–µ—Е–Њ–і–Њ–Љ —Б–Њ—Б—В–Њ—П–љ–Є—П –њ–∞—Ж–Є–µ–љ—В–∞ –Њ—В¬†–љ–∞—А—Г—И–µ–љ–Є—П —В–Њ–ї–µ—А–∞–љ—В–љ–Њ—Б—В–Є –Ї¬†–≥–ї—О–Ї–Њ–Ј–µ –Ї¬†—А–∞–Ј–≤–Є—В–Є—О –°–Ф, —З—В–Њ —В–∞–Ї–ґ–µ —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞–µ—В—Б—П –љ–∞—А–∞—Б—В–∞—О—Й–Є–Љ –≤–ї–Є—П–љ–Є–µ–Љ —Б–Њ—Б—Г–і–Є—Б—В—Л—Е —Д–∞–Ї—В–Њ—А–Њ–≤ —А–Є—Б–Ї–∞, —В–∞–Ї–Є—Е –Ї–∞–Ї –∞—А—В–µ—А–Є–Њ—Б–Ї–ї–µ—А–Њ–Ј –Є¬†–∞—А—В–µ—А–Є–∞–ї—М–љ–∞—П –≥–Є–њ–µ—А—В–µ–љ–Ј–Є—П. –£¬†–ї–Є—Ж –њ–Њ–ґ–Є–ї–Њ–≥–Њ –≤–Њ–Ј—А–∞—Б—В–∞ –≤–µ—А–Њ—П—В–љ–Њ—Б—В—М –Ї–Њ–≥–љ–Є—В–Є–≤–љ–Њ–≥–Њ —Б–љ–Є–ґ–µ–љ–Є—П –≤—Л—И–µ, —З—В–Њ –Њ–±—К—П—Б–љ—П–µ—В—Б—П –≤–Њ–Ј—А–∞—Б—В–љ—Л–Љ–Є –Є–Ј–Љ–µ–љ–µ–љ–Є—П–Љ–Є –≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ [6, 18, 19].
–†–Є—Б–Ї —А–∞–Ј–≤–Є—В–Є—П –Ф–≠ –љ–µ¬†–Ј–∞–≤–Є—Б–Є—В –Њ—В¬†–њ–Њ–ї–∞, —Н—В–љ–Є—З–µ—Б–Ї–Њ–є –њ—А–Є–љ–∞–і–ї–µ–ґ–љ–Њ—Б—В–Є. –Ю–і–љ–∞–Ї–Њ –Њ–љ –њ–Њ–≤—Л—И–∞–µ—В—Б—П –њ—А–Є —Г–≤–µ–ї–Є—З–µ–љ–Є–Є –і–ї–Є—В–µ–ї—М–љ–Њ—Б—В–Є –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є—П, –љ–µ–∞–і–µ–Ї–≤–∞—В–љ–Њ–Љ –Ї–Њ–љ—В—А–Њ–ї–µ –≥–Є–њ–µ—А–≥–ї–Є–Ї–µ–Љ–Є–Є, –∞—Б—Б–Њ—Ж–Є–Є—А–Њ–≤–∞–љ–љ–Њ–Љ —Б¬†–±–Њ–ї–µ–µ –Ј–љ–∞—З–Є—В–µ–ї—М–љ—Л–Љ –Љ–Є–Ї—А–Њ–≤–∞—Б–Ї—Г–ї—П—А–љ—Л–Љ –њ–Њ—А–∞–ґ–µ–љ–Є–µ–Љ, –њ–Њ–≤—Л—И–µ–љ–љ—Л–Љ —Г—А–Њ–≤–љ–µ–Љ –≥–ї–Є–Ї–Є—А–Њ–≤–∞–љ–љ–Њ–≥–Њ –≥–µ–Љ–Њ–≥–ї–Њ–±–Є–љ–∞, –Њ—В—А–∞–ґ–∞—О—Й–µ–≥–Њ —Б—В–µ–њ–µ–љ—М –њ–µ—А—Б–Є—Б—В–Є—А–Њ–≤–∞–љ–Є—П –≥–Є–њ–µ—А–≥–ї–Є–Ї–µ–Љ–Є–Є –≤¬†—В–µ—З–µ–љ–Є–µ –і–љ—П.
–Ъ–Њ–љ–≤–µ—А—Б–Є—П —Г–Љ–µ—А–µ–љ–љ—Л—Е –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л—Е —А–∞—Б—Б—В—А–Њ–є—Б—В–≤ –≤¬†–і–µ–Љ–µ–љ—Ж–Є—О –Є¬†–њ—А–Є—Б–Њ–µ–і–Є–љ–µ–љ–Є–µ –Ї–Њ–Љ–Њ—А–±–Є–і–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є
–†–∞–Ј–≤–Є—В–Є–µ –і–µ–Љ–µ–љ—Ж–Є–Є, —А–Є—Б–Ї –Ї–Њ—В–Њ—А–Њ–є –њ—А–Є –Ф–≠ –њ–Њ–≤—Л—И–µ–љ, –≤–µ—А–Њ—П—В–љ–Њ, —Б—В–∞–љ–Њ–≤–Є—В—Б—П –≤–Њ–Ј–Љ–Њ–ґ–љ—Л–Љ –љ–µ¬†–≤ —А–∞–Љ–Ї–∞—Е —Б–∞–Љ–Њ–є –Ф–≠, –∞ –≤¬†—А–µ–Ј—Г–ї—М—В–∞—В–µ –њ—А–Є—Б–Њ–µ–і–Є–љ–µ–љ–Є—П –Ї–Њ–Љ–Њ—А–±–Є–і–љ—Л—Е –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є¬†вАУ –С–Р –Є/–Є–ї–Є –≤—Л—А–∞–ґ–µ–љ–љ–Њ–є —Ж–µ—А–µ–±—А–Њ–≤–∞—Б–Ї—Г–ї—П—А–љ–Њ–є –њ–∞—В–Њ–ї–Њ–≥–Є–Є. –Ъ–∞–Ї —А–∞–Ј–≤–Є—В–Є–µ –Є–љ—Б—Г–ї—М—В–∞, —В–∞–Ї –Є¬†—Е—А–Њ–љ–Є—З–µ—Б–Ї–∞—П —Ж–µ—А–µ–±—А–Њ–≤–∞—Б–Ї—Г–ї—П—А–љ–∞—П –њ–∞—В–Њ–ї–Њ–≥–Є—П —Г—Б—Г–≥—Г–±–ї—П—О—В –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л–є —Б—В–∞—В—Г—Б –њ–∞—Ж–Є–µ–љ—В–∞ —Б¬†–Ф–≠. –Э–∞–ї–Є—З–Є–µ –љ–µ–Љ—Л—Е –Є–љ—Д–∞—А–Ї—В–Њ–≤ –Љ–Њ–Ј–≥–∞ —Г¬†–±–Њ–ї—М–љ—Л—Е –°–Ф —Г–і–≤–∞–Є–≤–∞–µ—В —А–Є—Б–Ї —А–∞–Ј–≤–Є—В–Є—П –і–µ–Љ–µ–љ—Ж–Є–Є –≤¬†—В–µ—З–µ–љ–Є–µ —З–µ—В—Л—А–µ—Е –ї–µ—В [3].
–Я—А–µ–і–Є–Ї—В–Њ—А–∞–Љ–Є –Ї–Њ–љ–≤–µ—А—Б–Є–Є —Г–Љ–µ—А–µ–љ–љ—Л—Е –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л—Е —А–∞—Б—Б—В—А–Њ–є—Б—В–≤ –≤¬†–і–µ–Љ–µ–љ—Ж–Є—О –Љ–Њ–≥—Г—В –≤—Л—Б—В—Г–њ–∞—В—М –≥–µ–љ–µ—В–Є—З–µ—Б–Ї–∞—П –њ—А–µ–і—А–∞—Б–њ–Њ–ї–Њ–ґ–µ–љ–љ–Њ—Б—В—М, –љ–∞–њ—А–Є–Љ–µ—А –∞–ї–ї–µ–ї—М –Р–†–Ю–Х4, –∞ —В–∞–Ї–ґ–µ –і–µ–њ—А–µ—Б—Б–Є—П, –Ї–Њ—В–Њ—А–∞—П –Љ–Њ–ґ–µ—В –Њ—В—А–∞–ґ–∞—В—М –Ї–∞–Ї –њ—Б–Є—Е–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –њ—А–Њ–±–ї–µ–Љ—Л –њ–∞—Ж–Є–µ–љ—В–Њ–≤, —В–∞–Ї –Є¬†–љ–µ–є—А–Њ–Љ–µ–і–Є–∞—В–Њ—А–љ—Л–µ –Є¬†—Б—В—А—Г–Ї—В—Г—А–љ—Л–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П –≤¬†–≤–µ—Й–µ—Б—В–≤–µ –≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞.
–Я–Њ¬†–љ–µ–Ї–Њ—В–Њ—А—Л–Љ –і–∞–љ–љ—Л–Љ, —Г¬†–±–Њ–ї—М–љ—Л—Е –°–Ф —З–∞—Й–µ –і–Є–∞–≥–љ–Њ—Б—В–Є—А—Г–µ—В—Б—П —Б–Љ–µ—И–∞–љ–љ–∞—П —Д–Њ—А–Љ–∞ –і–µ–Љ–µ–љ—Ж–Є–Є. –Ъ–Њ–Љ–±–Є–љ–∞—Ж–Є—П —Ж–µ—А–µ–±—А–Њ–≤–∞—Б–Ї—Г–ї—П—А–љ–Њ–≥–Њ –Є¬†–і–µ–≥–µ–љ–µ—А–∞—В–Є–≤–љ–Њ–≥–Њ –њ—А–Њ—Ж–µ—Б—Б–Њ–≤ —Б–Њ–њ—А–Њ–≤–Њ–ґ–і–∞–µ—В—Б—П –±–Њ–ї–µ–µ —В—П–ґ–µ–ї–Њ–є —Н–љ–і–Њ—В–µ–ї–Є–∞–ї—М–љ–Њ–є –і–Є—Б—Д—Г–љ–Ї—Ж–Є–µ–є, –љ–∞—А—Г—И–µ–љ–Є–µ–Љ —Ж–µ–ї–Њ—Б—В–љ–Њ—Б—В–Є –≥–µ–Љ–∞—В–Њ—Н–љ—Ж–µ—Д–∞–ї–Є—З–µ—Б–Ї–Њ–≥–Њ –±–∞—А—М–µ—А–∞, –±–Њ–ї–µ–µ —В—П–ґ–µ–ї—Л–Љ –Њ–Ї–Є—Б–ї–Є—В–µ–ї—М–љ—Л–Љ —Б—В—А–µ—Б—Б–Њ–Љ, –±–Њ–ї–µ–µ –Є–љ—В–µ–љ—Б–Є–≤–љ—Л–Љ –љ–∞–Ї–Њ–њ–ї–µ–љ–Є–µ–Љ –±–µ—В–∞-–∞–Љ–Є–ї–Њ–Є–і–∞ –Є¬†–љ–µ–є—А–Њ–≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ—Л–Љ –њ—А–Њ—Ж–µ—Б—Б–Њ–Љ. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –њ—А–Є —Б–Њ—З–µ—В–∞–љ–Є–Є –С–Р –Є¬†–°–Ф –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ —А–∞–Ј–≤–Є—В–Є–µ —Ж–µ—А–µ–±—А–∞–ї—М–љ–Њ–є –∞–Љ–Є–ї–Њ–Є–і–љ–Њ–є –∞–љ–≥–Є–Њ–њ–∞—В–Є–Є. –°–Ф —Г—Е—Г–і—И–∞–µ—В –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л–є —Б—В–∞—В—Г—Б –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†—А–∞–Ј–≤–Є–≤–∞—О—Й–µ–є—Б—П –С–Р. –Ю–і–љ–∞–Ї–Њ –љ–µ¬†–і–Њ–Ї–∞–Ј–∞–љ–Њ, —З—В–Њ –°–Ф —Г—Б—Г–≥—Г–±–ї—П–µ—В –њ–∞—В–Њ–Љ–Њ—А—Д–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є–µ –Є–Ј–Љ–µ–љ–µ–љ–Є—П, —Б–≤–Њ–є—Б—В–≤–µ–љ–љ—Л–µ –С–Р.
–Ы–µ—З–µ–љ–Є–µ –і–Є–∞–±–µ—В–Є—З–µ—Б–Ї–Њ–є¬†—Н–љ—Ж–µ—Д–∞–ї–Њ–њ–∞—В–Є–Є
–Ы–µ—З–µ–љ–Є–µ –Ф–≠ –і–Њ–ї–ґ–љ–Њ –≤–Ї–ї—О—З–∞—В—М –∞–і–µ–Ї–≤–∞—В–љ—Г—О —Д–Є–Ј–Є—З–µ—Б–Ї—Г—О –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –Є¬†–і–Є–µ—В—Г, –Ї–Њ—А—А–µ–Ї—Ж–Є—О –Є¬†—В—Й–∞—В–µ–ї—М–љ—Л–є –Ї–Њ–љ—В—А–Њ–ї—М —Г—А–Њ–≤–љ—П –≥–ї–Є–Ї–µ–Љ–Є–Є –Є¬†—Б–Њ—Б—Г–і–Є—Б—В—Л—Е —Д–∞–Ї—В–Њ—А–Њ–≤ —А–Є—Б–Ї–∞ [13, 20вАУ22].
–Я—А–µ–њ–∞—А–∞—В—Л –і–ї—П –ї–µ—З–µ–љ–Є—П –Ф–≠ –і–Њ–ї–ґ–љ—Л –±—Л—В—М –љ–∞–њ—А–∞–≤–ї–µ–љ—Л –љ–∞¬†—Г—Б–Є–ї–µ–љ–Є–µ –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є –Є–љ—Б—Г–ї–Є–Ј–Є–љ–∞, –њ–Њ–≤—Л—И–µ–љ–Є–µ —З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ–Њ—Б—В–Є —А–µ—Ж–µ–њ—В–Њ—А–Њ–≤ –Ї¬†–Є–љ—Б—Г–ї–Є–љ—Г, —З—В–Њ –Љ–Њ–ґ–µ—В —Б–њ–Њ—Б–Њ–±—Б—В–≤–Њ–≤–∞—В—М –њ–Њ–≤—Л—И–µ–љ–Є—О –Ї–ї–Є—А–µ–љ—Б–∞ –±–µ—В–∞-–∞–Љ–Є–ї–Њ–Є–і–∞. –Ь–Њ–і—Г–ї—П—В–Њ—А—Л –Є–љ—Б—Г–ї–Є–Ј–Є–љ–∞ –≤¬†—Д–Њ—А–Љ–µ –љ–µ–±–Њ–ї—М—И–Є—Е –њ–µ–њ—В–Є–і–Њ–≤ –Љ–Њ–≥—Г—В —Г—Б–Є–ї–Є—В—М –≥–Є–і—А–Њ–ї–Є–Ј –±–µ—В–∞-–∞–Љ–Є–ї–Њ–Є–і–∞, –љ–µ¬†–њ–Њ–≤—Л—И–∞—П —А–∞—Б–њ–∞–і —Б–∞–Љ–Њ–≥–Њ –Є–љ—Б—Г–ї–Є–љ–∞.
–Я—А–Є–Љ–µ–љ–µ–љ–Є–µ –њ–Є–Њ–≥–ї–Є—В–∞–Ј–Њ–љ–∞ вАУ –∞–≥–Њ–љ–Є—Б—В–∞ —А–µ—Ж–µ–њ—В–Њ—А–∞ –њ—А–Њ–ї–Є—Д–µ—А–∞—В–Њ—А–∞ –њ–µ—А–Њ–Ї—Б–Є—Б–Њ–Љ –≥–∞–Љ–Љ–∞ вАУ –њ—А–Є–≤–Њ–і–Є—В –Ї¬†—Г–ї—Г—З—И–µ–љ–Є—О —З—Г–≤—Б—В–≤–Є—В–µ–ї—М–љ–Њ—Б—В–Є –Ї¬†–Є–љ—Б—Г–ї–Є–љ—Г. –°–Є—Б—В–µ–Љ–љ–Њ–µ –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є–µ –њ—А–µ–њ–∞—А–∞—В–Њ–≤ –Є–љ—Б—Г–ї–Є–љ–∞ –і–ї—П –ї–µ—З–µ–љ–Є—П –Ј–∞–±–Њ–ї–µ–≤–∞–љ–Є–є –≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ –≤–Њ–Ј–Љ–Њ–ґ–љ–Њ, —В–∞–Ї –Ї–∞–Ї –Є–љ—Б—Г–ї–Є–љ —Б–њ–Њ—Б–Њ–±–µ–љ –њ—А–Њ–љ–Є–Ї–∞—В—М —З–µ—А–µ–Ј –≥–µ–Љ–∞—В–Њ—Н–љ—Ж–µ—Д–∞–ї–Є—З–µ—Б–Ї–Є–є –±–∞—А—М–µ—А —Б¬†–њ–Њ–Љ–Њ—Й—М—О —Б–њ–µ—Ж–Є–∞–ї—М–љ–Њ–є —В—А–∞–љ—Б–њ–Њ—А—В–љ–Њ–є —Б–Є—Б—В–µ–Љ—Л. –Ю–і–љ–∞–Ї–Њ –Њ–љ –і–Њ–ї–ґ–µ–љ –≤–≤–Њ–і–Є—В—М—Б—П –≤¬†–Ї–Њ–ї–Є—З–µ—Б—В–≤–∞—Е, –љ–µ¬†–≤—Л–Ј—Л–≤–∞—О—Й–Є—Е –≥–Є–њ–Њ–≥–ї–Є–Ї–µ–Љ–Є—О.
–Ш–љ—В—А–∞–љ–∞–Ј–∞–ї—М–љ–Њ–µ –≤–≤–µ–і–µ–љ–Є–µ –Є–љ—Б—Г–ї–Є–љ–∞, –њ—А–Њ–љ–Є–Ї–∞—О—Й–µ–≥–Њ –≤¬†–Љ–Њ–Ј–≥ —З–µ—А–µ–Ј —Б–Є—Б—В–µ–Љ—Г –Њ–±–Њ–љ—П—В–µ–ї—М–љ–Њ–≥–Њ –Є¬†—В—А–Њ–є–љ–Є—З–љ–Њ–≥–Њ –љ–µ—А–≤–Њ–≤, –Љ–Њ–ґ–µ—В —Б—В–∞—В—М –∞–ї—М—В–µ—А–љ–∞—В–Є–≤–Њ–є —Б–Є—Б—В–µ–Љ–љ–Њ–Љ—Г –≤–≤–µ–і–µ–љ–Є—О. –≠–Ї—Б–њ–µ—А–Є–Љ–µ–љ—В–∞–ї—М–љ—Л–µ –і–∞–љ–љ—Л–µ —Б–≤–Є–і–µ—В–µ–ї—М—Б—В–≤—Г—О—В, —З—В–Њ –Є–љ—В—А–∞–љ–∞–Ј–∞–ї—М–љ–Њ–µ –≤–≤–µ–і–µ–љ–Є–µ –Є–љ—Б—Г–ї–Є–љ–∞ —Б–њ–Њ—Б–Њ–±–љ–Њ –њ–Њ–ї–Њ–ґ–Є—В–µ–ї—М–љ–Њ –≤–ї–Є—П—В—М –љ–∞¬†–Ї–Њ–≥–љ–Є—В–Є–≤–љ–Њ–µ —Б–љ–Є–ґ–µ–љ–Є–µ, —Б—В–µ–њ–µ–љ—М –∞—В—А–Њ—Д–Є–Є –≥–Њ–ї–Њ–≤–љ–Њ–≥–Њ –Љ–Њ–Ј–≥–∞ –Є¬†–Є–Ј–Љ–µ–љ–µ–љ–Є—П –±–µ–ї–Њ–≥–Њ –≤–µ—Й–µ—Б—В–≤–∞, –љ–Њ¬†–ї–Є—И—М –њ—А–Є —Г—Б–ї–Њ–≤–Є–Є –Њ—В–љ–Њ—Б–Є—В–µ–ї—М–љ–Њ–≥–Њ –і–µ—Д–Є—Ж–Є—В–∞ –Є–љ—Б—Г–ї–Є–љ–∞, —З—В–Њ –Љ–Њ–ґ–µ—В –љ–∞–±–ї—О–і–∞—В—М—Б—П –Ї–∞–Ї –њ—А–Є –°–Ф 1¬†—В–Є–њ–∞, —В–∞–Ї –Є¬†–њ—А–Є –°–Ф 2 —В–Є–њ–∞ [4, 6, 13, 14, 20].
–Т–∞–ґ–µ–љ –њ–Њ–Є—Б–Ї —Н—Д—Д–µ–Ї—В–Є–≤–љ—Л—Е –Є¬†–±–µ–Ј–Њ–њ–∞—Б–љ—Л—Е –њ—А–µ–њ–∞—А–∞—В–Њ–≤, –њ—А–Њ—В–Є–≤–Њ–і–µ–є—Б—В–≤—Г—О—Й–Є—Е –Њ–Ї–Є—Б–ї–Є—В–µ–ї—М–љ–Њ–Љ—Г —Б—В—А–µ—Б—Б—Г, –љ–µ–є—А–Њ–≤–Њ—Б–њ–∞–ї–Є—В–µ–ї—М–љ–Њ–Љ—Г –њ—А–Њ—Ж–µ—Б—Б—Г, –∞ —В–∞–Ї–ґ–µ –Љ–Њ–і—Г–ї–Є—А—Г—О—Й–Є—Е –≥–ї—Г—В–∞–Љ–∞—В–µ—А–≥–Є—З–µ—Б–Ї—Г—О, —Е–Њ–ї–Є–љ–µ—А–≥–Є—З–µ—Б–Ї—Г—О –Є¬†–Ї–∞—В–µ—Е–Њ–ї–∞–Љ–Є–љ–µ—А–≥–Є—З–µ—Б–Ї—Г—О –њ–µ—А–µ–і–∞—З—Г.
–Я–µ—А—Б–њ–µ–Ї—В–Є–≤—Л –Љ—Г–ї—М—В–Є–Љ–Њ–і–∞–ї—М–љ–Њ–є¬†—В–µ—А–∞–њ–Є–Є –і–Є–∞–±–µ—В–Є—З–µ—Б–Ї–Њ–є —Н–љ—Ж–µ—Д–∞–ї–Њ–њ–∞—В–Є–Є
–Ґ–µ—А–∞–њ–Є—П, –Њ–і–љ–Њ–≤—А–µ–Љ–µ–љ–љ–Њ –љ–∞–њ—А–∞–≤–ї–µ–љ–љ–∞—П –љ–∞¬†–љ–µ—Б–Ї–Њ–ї—М–Ї–Њ –Ї–ї—О—З–µ–≤—Л—Е –њ–∞—В–Њ—Д–Є–Ј–Є–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Є—Е –Ј–≤–µ–љ—М–µ–≤ –Ф–≠, –њ–Њ-–≤–Є–і–Є–Љ–Њ–Љ—Г, —П–≤–ї—П–µ—В—Б—П –љ–∞–Є–±–Њ–ї–µ–µ –њ–µ—А—Б–њ–µ–Ї—В–Є–≤–љ–Њ–є. –Т¬†—Н—В–Њ–є —Б–≤—П–Ј–Є –њ—А–µ–і—Б—В–∞–≤–ї—П–µ—В—Б—П —Ж–µ–ї–µ—Б–Њ–Њ–±—А–∞–Ј–љ—Л–Љ –њ—А–Є–Љ–µ–љ–µ–љ–Є–µ –Ї–Њ–Љ–±–Є–љ–∞—Ж–Є–є –њ—А–µ–њ–∞—А–∞—В–Њ–≤ –Є–ї–Є –Љ–љ–Њ–≥–Њ–Ї–Њ–Љ–њ–Њ–љ–µ–љ—В–љ—Л—Е –њ—А–µ–њ–∞—А–∞—В–Њ–≤. –Я—А–Є–Љ–µ—А–Њ–Љ –њ–Њ—Б–ї–µ–і–љ–µ–≥–Њ –Љ–Њ–ґ–µ—В —Б–ї—Г–ґ–Є—В—М –Р–Ї—В–Њ–≤–µ–≥–Є–љ [12, 14, 22, 23].
–Р–Ї—В–Њ–≤–µ–≥–Є–љ вАУ –і–µ–њ—А–Њ—В–µ–Є–љ–Є–Ј–Є—А–Њ–≤–∞–љ–љ—Л–є –≥–µ–Љ–Њ–і–µ—А–Є–≤–∞—В, –њ–Њ–ї—Г—З–∞–µ–Љ—Л–є –Є–Ј¬†–Ї—А–Њ–≤–Є —В–µ–ї—П—В –њ—Г—В–µ–Љ —Г–ї—М—В—А–∞—Д–Є–ї—М—В—А–∞—Ж–Є–Є, –≤¬†—Б–Њ—Б—В–∞–≤ –Ї–Њ—В–Њ—А–Њ–≥–Њ –≤—Е–Њ–і—П—В –Њ–Ї–Њ–ї–Њ 200 –љ–Є–Ј–Ї–Њ–Љ–Њ–ї–µ–Ї—Г–ї—П—А–љ—Л—Е —Б–Њ–µ–і–Є–љ–µ–љ–Є–є –≤–µ—Б–Њ–Љ –і–Њ¬†5000 –і–∞–ї—М—В–Њ–љ. –Р–Ї—В–Њ–≤–µ–≥–Є–љ —Б–Њ–і–µ—А–ґ–Є—В –Є–љ–Њ–Ј–Є—В–Њ–ї—Д–Њ—Б—Д–Њ–Њ–ї–Є–≥–Њ—Б–∞—Е–∞—А–Є–і—Л, –Ї–Њ—В–Њ—А—Л–µ –∞–Ї—В–Є–≤–Є—А—Г—О—В —В—А–∞–љ—Б–њ–Њ—А—В –≥–ї—О–Ї–Њ–Ј—Л –Є¬†—Б—В–Є–Љ—Г–ї–Є—А—Г—О—В –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –љ–µ–Ї–Њ—В–Њ—А—Л—Е —Д–µ—А–Љ–µ–љ—В–Њ–≤, –≤–Ї–ї—О—З–∞—П –њ–Є—А—Г–≤–∞—В–і–µ–≥–Є–і—А–Њ–≥–µ–љ–∞–Ј—Г вАУ –Ї–ї—О—З–µ–≤–Њ–є —Д–µ—А–Љ–µ–љ—В —Ж–Є–Ї–ї–∞ –Ъ—А–µ–±—Б–∞. –Ш–љ—Б—Г–ї–Є–љ–љ–µ–Ј–∞–≤–Є—Б–Є–Љ–∞—П –∞–Ї—В–Є–≤–∞—Ж–Є—П –њ–µ—А–µ–љ–Њ—Б—З–Є–Ї–Њ–≤ –≥–ї—О–Ї–Њ–Ј—Л –Љ–Њ–ґ–µ—В –Њ—Б–ї–∞–±–ї—П—В—М –Љ–µ—В–∞–±–Њ–ї–Є—З–µ—Б–Ї–Є–µ –њ–Њ—Б–ї–µ–і—Б—В–≤–Є—П –Є–љ—Б—Г–ї–Є–љ–Њ—А–µ–Ј–Є—Б—В–µ–љ—В–љ–Њ—Б—В–Є, —П–≤–ї—П—О—Й–µ–є—Б—П –Њ–і–љ–Є–Љ –Є–Ј¬†—Д–∞–Ї—В–Њ—А–Њ–≤ –∞–ї—М—Ж–≥–µ–є–Љ–µ—А–Њ–≤—Б–Ї–Є—Е –Є–Ј–Љ–µ–љ–µ–љ–Є–є –≤¬†–Љ–Њ–Ј–≥–µ.
–£—Б—В–∞–љ–Њ–≤–ї–µ–љ–Њ, —З—В–Њ –Р–Ї—В–Њ–≤–µ–≥–Є–љ —Г–≤–µ–ї–Є—З–Є–≤–∞–µ—В —Г—В–Є–ї–Є–Ј–∞—Ж–Є—О –Ї–Є—Б–ї–Њ—А–Њ–і–∞, —З—В–Њ –њ—А–Є–≤–Њ–і–Є—В –Ї¬†—Г–≤–µ–ї–Є—З–µ–љ–Є—О —Б–Є–љ—В–µ–Ј–∞ –∞–і–µ–љ–Њ–Ј–Є–љ—В—А–Є—Д–Њ—Б—Д–∞—В–∞ –Є¬†–Ї—А–µ–∞—В–Є–љ—Д–Њ—Б—Д–∞—В–∞ –Є, –Ї–∞–Ї —Б–ї–µ–і—Б—В–≤–Є–µ, –Ј–∞—Й–Є—В–µ –Ї–ї–µ—В–Њ–Ї –Њ—В¬†–≥–Є–њ–Њ–Ї—Б–Є—З–µ—Б–Ї–Њ–≥–Њ –њ–Њ–≤—А–µ–ґ–і–µ–љ–Є—П. –Р–Ї—В–Њ–≤–µ–≥–Є–љ –Њ–Ї–∞–Ј—Л–≤–∞–µ—В –љ–µ–є—А–Њ–њ—А–Њ—В–µ–Ї—В–Є–≤–љ—Л–є –Є¬†–љ–µ–є—А–Њ—В—А–Њ—Д–Є—З–µ—Б–Ї–Є–є —Н—Д—Д–µ–Ї—В—Л, —Б–њ–Њ—Б–Њ–±—Б—В–≤—Г—П —В–Њ—А–Љ–Њ–ґ–µ–љ–Є—О –∞–њ–Њ–њ—В–Њ–Ј–∞ (–Ј–∞¬†—Б—З–µ—В –±–ї–Њ–Ї–Є—А–Њ–≤–∞–љ–Є—П –Ї–∞—Б–њ–∞–Ј—Л-3), —Г–≤–µ–ї–Є—З–µ–љ–Є—О —З–Є—Б–ї–µ–љ–љ–Њ—Б—В–Є –љ–µ–є—А–Њ–љ–Њ–≤ –Є¬†—Б–Є–љ–∞–њ—В–Є—З–µ—Б–Ї–Є—Е —Б–≤—П–Ј–µ–є. –Я—А–µ–њ–∞—А–∞—В —Б–њ–Њ—Б–Њ–±–µ–љ –Љ–Њ–і—Г–ї–Є—А–Њ–≤–∞—В—М –∞–Ї—В–Є–≤–љ–Њ—Б—В—М –љ—Г–Ї–ї–µ–∞—А–љ–Њ–≥–Њ (—П–і–µ—А–љ–Њ–≥–Њ) —Д–∞–Ї—В–Њ—А–∞ ќЇB, –Є–≥—А–∞—О—Й–µ–≥–Њ –≤–∞–ґ–љ—Г—О —А–Њ–ї—М –≤¬†—А–µ–≥—Г–ї—П—Ж–Є–Є –њ—А–Њ—Ж–µ—Б—Б–Њ–≤ –∞–њ–Њ–њ—В–Њ–Ј–∞ –Є¬†–≤–Њ—Б–њ–∞–ї–µ–љ–Є—П. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, –Њ–љ —Б–њ–Њ—Б–Њ–±–µ–љ —Г–ї—Г—З—И–∞—В—М –Љ–Є–Ї—А–Њ—Ж–Є—А–Ї—Г–ї—П—Ж–Є—О –≤¬†—В–Ї–∞–љ—П—Е, –њ–Њ–Ј–Є—В–Є–≤–љ–Њ –≤–Њ–Ј–і–µ–є—Б—В–≤—Г—П –љ–∞¬†—Н–љ–і–Њ—В–µ–ї–Є–є —Б–Њ—Б—Г–і–Њ–≤ [14, 16, 24вАУ28].
–Т¬†–њ–ї–∞—Ж–µ–±–Њ–Ї–Њ–љ—В—А–Њ–ї–Є—А—Г–µ–Љ—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П—Е –њ—А–Њ–і–µ–Љ–Њ–љ—Б—В—А–Є—А–Њ–≤–∞–љ–∞ —Б–њ–Њ—Б–Њ–±–љ–Њ—Б—В—М –њ—А–µ–њ–∞—А–∞—В–∞ –Р–Ї—В–Њ–≤–µ–≥–Є–љ —Г–ї—Г—З—И–∞—В—М –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л–µ —Д—Г–љ–Ї—Ж–Є–Є –Є¬†—Б—В–µ–њ–µ–љ—М –њ–Њ–≤—Б–µ–і–љ–µ–≤–љ–Њ–є –∞–Ї—В–Є–≤–љ–Њ—Б—В–Є —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†—Б–Њ—Б—Г–і–Є—Б—В–Њ–є –і–µ–Љ–µ–љ—Ж–Є–µ–є –Є¬†–С–Р [27, 29вАУ32].
–Т¬†—А—П–і–µ –Њ—В–Ї—А—Л—В—Л—Е –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–є –њ–Њ–Ї–∞–Ј–∞–љ–Њ –њ–Њ–ї–Њ–ґ–Є—В–µ–ї—М–љ–Њ–µ –≤–Њ–Ј–і–µ–є—Б—В–≤–Є–µ –њ—А–µ–њ–∞—А–∞—В–∞ –Р–Ї—В–Њ–≤–µ–≥–Є–љ –љ–∞¬†—Б–Њ—Б—В–Њ—П–љ–Є–µ –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л—Е —Д—Г–љ–Ї—Ж–Є–є —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–Ф–≠ [2, 14]. –Т¬†–Ї—А—Г–њ–љ–Њ–Љ –Љ–љ–Њ–≥–Њ—Ж–µ–љ—В—А–Њ–≤–Њ–Љ –њ–ї–∞—Ж–µ–±–Њ–Ї–Њ–љ—В—А–Њ–ї–Є—А—Г–µ–Љ–Њ–Љ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є–Є —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–°–Ф 2 —В–Є–њ–∞ –љ–∞¬†—Д–Њ–љ–µ –њ—А–Є–µ–Љ–∞ –њ—А–µ–њ–∞—А–∞—В–∞ –Р–Ї—В–Њ–≤–µ–≥–Є–љ –Њ—В–Љ–µ—З–µ–љ–Њ —Г–ї—Г—З—И–µ–љ–Є–µ —Б–Њ¬†—Б—В–Њ—А–Њ–љ—Л –Я–Э–° –Є¬†–њ—Б–Є—Е–Њ–ї–Њ–≥–Є—З–µ—Б–Ї–Њ–≥–Њ —Б–Њ—Б—В–Њ—П–љ–Є—П (–њ–Њ¬†—И–Ї–∞–ї–µ –Ї–∞—З–µ—Б—В–≤–∞ –ґ–Є–Ј–љ–Є)¬†[12].
–†–µ–Ј—Г–ї—М—В–∞—В—Л –љ–µ–і–∞–≤–љ–Њ –Ј–∞–≤–µ—А—И–Є–≤—И–µ–≥–Њ—Б—П –Љ–љ–Њ–≥–Њ—Ж–µ–љ—В—А–Њ–≤–Њ–≥–Њ —А–∞–љ–і–Њ–Љ–Є–Ј–Є—А–Њ–≤–∞–љ–љ–Њ–≥–Њ –Є—Б—Б–ї–µ–і–Њ–≤–∞–љ–Є—П ARTEMIDA, –≤¬†–Ї–Њ—В–Њ—А–Њ–Љ –Є–Ј—Г—З–∞–ї–∞—Б—М —Н—Д—Д–µ–Ї—В–Є–≤–љ–Њ—Б—В—М –њ—А–µ–њ–∞—А–∞—В–∞ —Г¬†–њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–њ–Њ—Б—В–Є–љ—Б—Г–ї—М—В–љ—Л–Љ–Є –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л–Љ–Є –љ–∞—А—Г—И–µ–љ–Є—П–Љ–Є –њ—А–Є –љ–∞–Ј–љ–∞—З–µ–љ–Є–Є –≤¬†–Њ—Б—В—А–Њ–Љ –њ–µ—А–Є–Њ–і–µ –Є–љ—Б—Г–ї—М—В–∞, –њ–Њ–Ї–∞–Ј–∞–ї–Є, —З—В–Њ —З–µ—А–µ–Ј —И–µ—Б—В—М –Љ–µ—Б—П—Ж–µ–≤ –њ—А–µ–њ–∞—А–∞—В–Њ–Љ –Р–Ї—В–Њ–≤–µ–≥–Є–љ –і–Њ—Б—В–Њ–≤–µ—А–љ–Њ —Г–ї—Г—З—И–∞—О—В—Б—П –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л–µ —Д—Г–љ–Ї—Ж–Є–Є –њ–Њ¬†—Б—А–∞–≤–љ–µ–љ–Є—О —Б¬†–њ—А–Є–µ–Љ–Њ–Љ –њ–ї–∞—Ж–µ–±–Њ. –Ъ—А–Њ–Љ–µ —В–Њ–≥–Њ, —Б–љ–Є–ґ–∞–µ—В—Б—П –Ї–Њ–ї–Є—З–µ—Б—В–≤–Њ –њ–∞—Ж–Є–µ–љ—В–Њ–≤ —Б¬†–і–Є–∞–≥–љ–Њ–Ј–Њ–Љ ¬Ђ–і–µ–Љ–µ–љ—Ж–Є—П¬ї. –£–Ї–∞–Ј–∞–љ–љ—Л–є —Н—Д—Д–µ–Ї—В –Њ—В–Љ–µ—З–∞–ї—Б—П –Ї–∞–Ї —З–µ—А–µ–Ј —И–µ—Б—В—М –Љ–µ—Б—П—Ж–µ–≤ —В–µ—А–∞–њ–Є–Є, —В–∞–Ї –Є¬†—З–µ—А–µ–Ј —И–µ—Б—В—М –Љ–µ—Б—П—Ж–µ–≤ –њ–Њ—Б–ї–µ–і—Г—О—Й–µ–≥–Њ –љ–∞–±–ї—О–і–µ–љ–Є—П [12].
–Ґ–∞–Ї–Є–Љ –Њ–±—А–∞–Ј–Њ–Љ, –Р–Ї—В–Њ–≤–µ–≥–Є–љ –Љ–Њ–ґ–µ—В –њ—А–Є–Љ–µ–љ—П—В—М—Б—П –Ї–∞–Ї –њ—А–Є –ї–µ–≥–Ї–Є—Е –Є¬†—Г–Љ–µ—А–µ–љ–љ—Л—Е –Ї–Њ–≥–љ–Є—В–Є–≤–љ—Л—Е –љ–∞—А—Г—И–µ–љ–Є—П—Е, —В–∞–Ї –Є¬†–љ–∞ —Б—В–∞–і–Є–Є –і–µ–Љ–µ–љ—Ж–Є–Є –≤¬†–≤–Є–і–µ –Љ–Њ–љ–Њ—В–µ—А–∞–њ–Є–Є –Є–ї–Є –≤¬†–Ї–Њ–Љ–±–Є–љ–∞—Ж–Є–Є —Б¬†–њ—А–µ–њ–∞—А–∞—В–∞–Љ–Є, –≤–Њ–Ј–і–µ–є—Б—В–≤—Г—О—Й–Є–Љ–Є –љ–∞¬†–љ–µ–є—А–Њ–Љ–µ–і–Є–∞—В–Њ—А–љ—Л–µ —Б–Є—Б—В–µ–Љ—Л. –£—З–Є—В—Л–≤–∞—П –і–Њ–Ј–Њ–Ј–∞–≤–Є—Б–Є–Љ—Л–є —Е–∞—А–∞–Ї—В–µ—А –і–µ–є—Б—В–≤–Є—П, –њ—А–µ–њ–∞—А–∞—В –љ–µ–Њ–±—Е–Њ–і–Є–Љ–Њ –њ—А–Є–Љ–µ–љ—П—В—М –≤¬†–∞–і–µ–Ї–≤–∞—В–љ—Л—Е –і–Њ–Ј–∞—Е: –≤–љ—Г—В—А–Є–≤–µ–љ–љ–Њ –Ї–∞–њ–µ–ї—М–љ–Њ –њ–Њ¬†0,4вАУ2¬†–≥ (–љ–∞¬†–Ї—Г—А—Б –і–Њ¬†20¬†–Є–љ—Д—Г–Ј–Є–є), –њ–µ—А–Њ—А–∞–ї—М–љ–Њ вАУ –њ–Њ¬†400 –Љ–≥ —В—А–Є —А–∞–Ј–∞ –≤¬†–і–µ–љ—М. –Ф–ї–Є—В–µ–ї—М–љ–Њ—Б—В—М –њ—А–Є–µ–Љ–∞ –Ј–∞–≤–Є—Б–Є—В –Њ—В¬†–≤—Л—А–∞–ґ–µ–љ–љ–Њ—Б—В–Є —Б–Є–Љ–њ—В–Њ–Љ–∞—В–Є–Ї–Є. –Т¬†—Б—А–µ–і–љ–µ–Љ –Њ–љ–∞ —Б–Њ—Б—В–∞–≤–ї—П–µ—В –Њ—В¬†—З–µ—В—Л—А–µ—Е вАУ —И–µ—Б—В–Є –љ–µ–і–µ–ї—М –і–Њ¬†—И–µ—Б—В–Є –Љ–µ—Б—П—Ж–µ–≤.
O.S. Levin, O.V. Babkina
Russian Medical Academy of Postgraduate Education
Contact person: Oleg Semyonovich Levin, oslevin@mail.ru
Impact of diabetes mellitus (DM) on nervous system is routinely associated with injury of peripheral nerves and nervous fibers. However, recently more interest was risen by dysfunction of the brain related to development of cognitive impairment known as diabetic encephalopathy (DE) implying slowly deteriorating cognitive impairment that usually does not reach (within DE) dementia, and caused by metabolic disorders directly triggered by DM.
A substrate of encephalopathy remains unclear, and its state, diagnostic criteria are still debated explaining why this term has been much rarely used in last years. DE develops primary to type 1 DM, which is related to impaired action of insulin and hyperglycemia, whereas during type 2 DM it is a consequence of vascular complications in substantial proportions of cases.
–£–≤–∞–ґ–∞–µ–Љ—Л–є –њ–Њ—Б–µ—В–Є—В–µ–ї—М uMEDp!
–£–≤–µ–і–Њ–Љ–ї—П–µ–Љ –Т–∞—Б –Њ —В–Њ–Љ, —З—В–Њ –Ј–і–µ—Б—М —Б–Њ–і–µ—А–ґ–Є—В—Б—П –Є–љ—Д–Њ—А–Љ–∞—Ж–Є—П, –њ—А–µ–і–љ–∞–Ј–љ–∞—З–µ–љ–љ–∞—П –Є—Б–Ї–ї—О—З–Є—В–µ–ї—М–љ–Њ –і–ї—П —Б–њ–µ—Ж–Є–∞–ї–Є—Б—В–Њ–≤ –Ј–і—А–∞–≤–Њ–Њ—Е—А–∞–љ–µ–љ–Є—П.
–Х—Б–ї–Є –Т—Л –љ–µ —П–≤–ї—П–µ—В–µ—Б—М —Б–њ–µ—Ж–Є–∞–ї–Є—Б—В–Њ–Љ –Ј–і—А–∞–≤–Њ–Њ—Е—А–∞–љ–µ–љ–Є—П, –∞–і–Љ–Є–љ–Є—Б—В—А–∞—Ж–Є—П –љ–µ –љ–µ—Б–µ—В –Њ—В–≤–µ—В—Б—В–≤–µ–љ–љ–Њ—Б—В–Є –Ј–∞ –≤–Њ–Ј–Љ–Њ–ґ–љ—Л–µ –Њ—В—А–Є—Ж–∞—В–µ–ї—М–љ—Л–µ –њ–Њ—Б–ї–µ–і—Б—В–≤–Є—П, –≤–Њ–Ј–љ–Є–Ї—И–Є–µ –≤ —А–µ–Ј—Г–ї—М—В–∞—В–µ —Б–∞–Љ–Њ—Б—В–Њ—П—В–µ–ї—М–љ–Њ–≥–Њ –Є—Б–њ–Њ–ї—М–Ј–Њ–≤–∞–љ–Є—П –Т–∞–Љ–Є –Є–љ—Д–Њ—А–Љ–∞—Ж–Є–Є —Б –њ–Њ—А—В–∞–ї–∞ –±–µ–Ј –њ—А–µ–і–≤–∞—А–Є—В–µ–ї—М–љ–Њ–є –Ї–Њ–љ—Б—Г–ї—М—В–∞—Ж–Є–Є —Б –≤—А–∞—З–Њ–Љ.
–Э–∞–ґ–Є–Љ–∞—П –љ–∞ –Ї–љ–Њ–њ–Ї—Г ¬Ђ–Т–Њ–є—В–Є¬ї, –Т—Л –њ–Њ–і—В–≤–µ—А–ґ–і–∞–µ—В–µ, —З—В–Њ —П–≤–ї—П–µ—В–µ—Б—М –≤—А–∞—З–Њ–Љ –Є–ї–Є —Б—В—Г–і–µ–љ—В–Њ–Љ –Љ–µ–і–Є—Ж–Є–љ—Б–Ї–Њ–≥–Њ –≤—Г–Ј–∞.