
Введение
В настоящее время во всех странах наблюдается рост заболеваемости сахарным диабетом (СД) и хронической болезнью почек (ХБП), которые часто сопутствуют друг другу [1–4]. Всего в мире насчитывается около 366 млн человек, страдающих СД. Подсчитано, что к 2030 г. число таких больных возрастет до 552 млн [5–7].
СД является ведущей причиной хронической почечной недостаточности (ХПН). Так, в США в 2005 г. СД было обусловлено 44% новых случаев ХПН [8]. По данным Национального института сахарного диабета, болезней пищеварительной системы и почек США (National Institute of Diabetes and Digestive and Kidney Disease), количество пациентов, получающих заместительную почечную терапию, в 12 раз выше среди больных СД, чем среди лиц без СД (133 на 100 тыс. населения и 11 на 100 тыс. населения соответственно) [9]. В 2003 г. в Японии было зарегистрировано более 237 тыс. пациентов, получающих заместительную почечную терапию, 41% из них составили больные с диабетической нефропатией на стадии ХПН [10].
Известно, что около 45% больных ХПН умирают, прежде чем начата заместительная почечная терапия [11]. В случае сочетанной патологии (ХБП и СД) этот показатель может быть гораздо выше. Выживаемость больных ХПН также зависит от наличия сердечно-сосудистых заболеваний [2, 3, 12], которые являются основной причиной смерти в этой группе пациентов [1–3, 11, 12].
Минерально-костные нарушения у пациентов с ХБП и СД
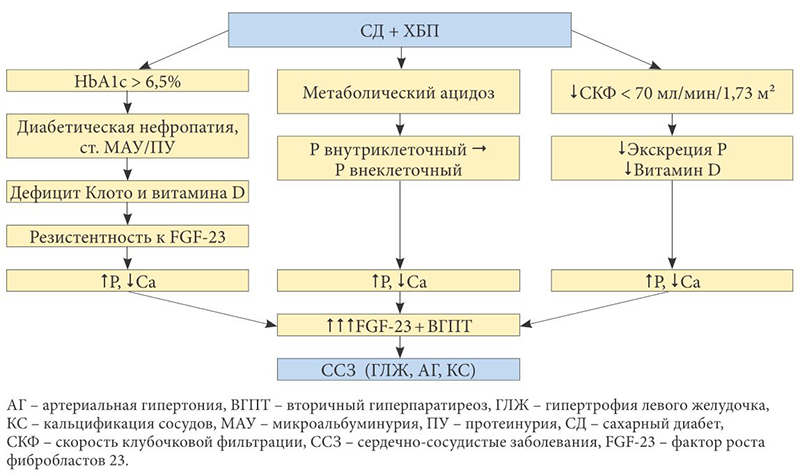
К сердечно-сосудистой заболеваемости и смертности среди пациентов с СД и ХБП могут привести минерально-костные нарушения [13] – системные нарушения минерального и костного метаболизма, обусловленные снижением почечной функции. Упрощенная схема развития минерально-костных нарушений у пациентов ХБП и СД представлена на рисунке 1.
Минерально-костные нарушения при ХБП – термин, утвержденный Инициативой по улучшению глобальных исходов заболеваний почек (Kidney Disease: Improving Global Outcomes, KDIGO). Новый термин заменил старое определение почечной остеодистрофии, которое не охватывало весь комплекс взаимодействий между Ca, P, паратиреоидным гормоном (ПТГ) и витамином D, нарушением костного метаболизма, минерализацией, объемом, линейным ростом или силой и кальцификацией сосудов и других мягких тканей [14, 15]. Минерально-костные нарушения у пациентов с ХБП определяют уже при скорости клубочковой фильтрации менее 70 мл/мин/1,73м² (II стадия ХБП) [16]. Эти нарушения могут приводить к развитию вторичного гиперпаратиреоза в результате ухудшения функции почек.
Костный метаболизм
Кость – орган-мишень многочисленных изменений, вызванных нарушением функции почек. В костях сосуществуют два основных процесса: резорбция и формирование ткани, направленные на поддержание гомеостаза кальция и фосфора, а также устранение микроповреждений кости и формирование скелета.
Изменения костного метаболизма в патогенезе минерально-костных нарушений у пациентов с ХБП и СД обусловлены:
- нарушением депонирования Р и Ca;
- нарушениями метаболизма глюкозы [17];
- действием инсулиноподобных факторов роста 1 и 2 типа;
- поддержанием кислотно-щелочного баланса путем абсорбции и высвобождения щелочных солей – буферов против изменения рН;
- накоплением ряда металлов (например, при применении соединений Al для гемодиализа в прошлом);
- эндокринной функцией – синтезом фактора роста фибробластов 23 (fibroblast growth factor-23, FGF-23), снижающего реабсорбцию фосфата почками [18], синтезом остеокальцина. Остеокальцин способствует регуляции гликемии, уменьшению висцеральных жировых отложений, повышению секреции и числа инсулинпродуцирующих клеток и чувствительности к инсулину [19].
Для минерально-костных нарушений при ХБП характерно несколько типов поражения скелета: с высоким обменом (высокий уровень ПТГ, с гиперактивными клетками кости и избыточной костной резорбцией), низким обменом (нормальный или низкий уровень ПТГ, адинамическая кость, небольшая активность клеток кости или ее отсутствие), смешанным обменом (черты обоих вышеуказанных типов без корреляции с уровнем ПТГ), остеопороз (наиболее часто у пожилых больных СД или как осложнение терапии кортикостероидами) и остеомаляция [15].
Фосфор
Начиная со II стадии ХБП при скорости клубочковой фильтрации менее 70 мл/мин/1,73м² [16] уменьшается экскреция фосфора почками. Это приводит к гиперфосфатемии (сывороточный P > 4,6 мг/дл), ингибированию 1-альфа-гидроксилазы и в дальнейшем к невозможности превращения витамина D в его активный метаболит – кальцитриол – в проксимальных почечных канальцах. Высокий уровень фосфора индуцирует повышение уровня FGF-23, синтезируемого остеоцитами, который оказывает фосфатурический эффект [18, 20], уменьшает всасывание фосфора в кишечнике, прямо воздействует на паращитовидные железы, стимулируя секрецию ПТГ, и подавляет образование кальцитриола, являясь его прямым антагонистом [21].
Уровень фосфора у пациентов с СД может быть повышен также из-за метаболического ацидоза и выраженного дефицита инсулина (гипергликемии), что обусловливает перемещение внутриклеточного фосфора во внеклеточную жидкость [22–24].
Гиперфосфатемия, которая запускает «порочный круг», связывающий все компоненты минерально-костных нарушений у пациентов с ХБП [25], повышение FGF-23 и ПТГ, кальцификацию сосудов [26, 27], признана независимым фактором риска сердечно-сосудистых заболеваний [28].
Кальций
Гомеостаз кальция является результатом равновесия следующих процессов: кишечного всасывания, обмена в костях, реабсорбции и выведения почками. Эти процессы регулируются ПТГ и кальцитриолом, которые повышают уровень Ca в крови.
Гипокальциемия характерна для пациентов с ХБП, особенно при СКФ менее 20 мл/мин/1,73м². Гипокальциемия индуцирует секрецию ПТГ, который повышает уровень циркулирующего Ca. ПТГ стимулирует активность остеокластов с высвобождением Ca и влияет на активный витамин D, усиливая всасывание Ca в кишечнике и реабсорбцию в почках [15, 16, 29].
Обмены кальция и фосфора тесно взаимосвязаны, поэтому для выявления минерально-костных нарушений у пациентов с ХБП рассматривается значение произведения Ca × P, но не уровни Ca или P в отдельности. У пациентов, получающих лечение диализом, произведение Ca × P должно быть < 5,5 ммоль²/л². Более высокие значения прямо коррелируют с кальцификацией митрального клапана и сосудов [27, 29]. В крупном европейском исследовании было показано, что уровень общего Ca > 2,75 ммоль/л и уровень Р > 1,78 ммоль/л повышает риск смерти на 70% и 32% соответственно. В то же время гипокальциемия не сопровождалась повышением частоты смертей [30].
Применение кальция и/или витамина D может улучшать показатели гликемии, уровень инсулина в крови и предупредить развитие СД. Проведенный метаанализ показал, что терапия витамином D и/или кальцием играет определенную роль в профилактике СД 2 типа, но только в группах высокого риска (с нарушенной толерантностью к глюкозе) [31].
Витамин D
Витамин D – прегормон, получаемый с пищей (10–20%) или синтезируемый в коже (воздействие ультрафиолета преобразует 7-дезоксихолестерол в холекальциферол – витамин D₃). Витамин D₃ проходит активацию в два этапа. Первый – в печени через 25-гидроксилирование с образованием 25-гидроксивитамина D, второй – в почках через 1-альфа-гидроксилирование с образованием активного метаболита 1,25-дигидроксивитамина D, или кальцитриола. Небольшое количество кальцитриола может быть получено через альтернативный путь, так как 1-альфа-гидроксилирование происходит во многих других органах (легких, толстой кишке, молочной железе или простате). Оптимальными значениями сывороточного 25-гидроксивитамина D признаны
40–80 нг/мл [32].
При диабетической нефропатии снижение уровня витамина D может быть обусловлено микро- и макроальбуминурией, протеинурией (почечные потери белка, связывающего витамин D), однако патогенез гиповитаминоза D до конца не ясен [33, 34].
Дефицит витамина D характерен для пациентов с ХБП и приводит к низкому всасыванию кальция в кишечнике, гипокальциемии и увеличению секреции ПТГ с развитием вторичного гиперпаратиреоза, который является независимым фактором риска смерти у пациентов с ХБП [2, 3].
Терапия витамином D направлена на коррекцию гипокальциемии и уменьшает проявления вторичного гиперпаратиреоза, но может осложняться гиперкальциемией и гиперфосфатемией за счет увеличения интестинального всасывания Ca и Р, так как гиперкальциемия и гиперфосфатемия прямо коррелируют с кальцификацией сосудов [35]. В этой связи лечение витамином D должно проводиться под систематическим контролем уровня витамина D, Ca, P и ПТГ.
Несмотря на то что кальцитриол опосредованно регулирует жесткость артериальной стенки, а высокое артериальное давление оказывает влияние на процессы кальцификации сосудов и сердечно-сосудистые осложнения, витамин D, согласно результатам метаанализа, не является независимым фактором риска сердечно-сосудистых заболеваний [36].
Парикальцитол – селективный агонист рецепторов витамина D – обладает плейотропным эффектом кальцитриола, и его применение ассоциировано с более выраженным подавлением секреции ПТГ, ингибированием синтеза ренина (уменьшением активности ренин-ангиотензин-альдостероновой системы и, как следствие, замедлением прогрессирования диабетической нефропатии), снижением протеинурии, активности воспаления, атеросклероза и кальцификации сосудов [36, 37].
Паратиреоидный гормон и фактор роста фибробластов 23
ПТГ и FGF-23 являются фосфатурическими гормонами. У здоровых лиц высокий уровень фосфатов в сыворотке индуцирует секрецию ПТГ и FGF-23, которые снижают реабсорбцию и увеличивают экскрецию фосфатов почками [28, 38].
ПТГ – пептидный гормон, секретируемый главными клетками паращитовидных желез, – обеспечивает системный гомеостаз кальция, действуя на основные органы-мишени – кости и почки. На кости ПТГ оказывает двойное действие: увеличивает число и активность остеобластов (анаболический эффект), а также активирует остеокласты (увеличивает резорбцию костной ткани). В почках ПТГ повышает 1-альфа-гидроксилирование, активируя продукцию кальцитриола, который усиливает кишечное всасывание Ca и P [39]. Таким образом, повышение уровня ПТГ увеличивает концентрацию внутриклеточного кальция, а также секрецию FGF-23, который подавляет образование кальцитриола и является его прямым антагонистом [38].
Секрецию ПТГ непосредственно регулируют ионизированный Ca, витамин D, P, Mg. Гипокальциемия и гиперфосфатемия стимулируют, а гиперкальциемия, витамин D и выраженная гипомагниемия тормозят образование ПТГ.
Сочетание СД и ХБП повышает риск метаболического ацидоза из-за снижения реабсорбции бикарбоната в почках. Ацидоз стимулирует образование ПТГ и костную резорбцию. Терапия натрия бикарбонатом может снижать уровень ПТГ до 20% [40], что имеет большое значение, поскольку вторичный гиперпаратиреоз осложняется кальцификацией мягких тканей, сердечных клапанов и сосудов (ПТГ стимулирует высвобождение Ca из костей и отложение в гладких мышечных клетках). Кроме того, уровень ПТГ прямо ассоциирован с артериальной гипертонией, гипертрофией левого желудочка и является независимым фактором риска сердечно-сосудистой заболеваемости и смертности [41].
FGF-23 – новый гормон среди признанных показателей (ПТГ и витамин D) минерально-костных нарушений у пациентов с ХБП. FGF-23 вырабатывается остеоцитами, и его основной функцией является стимуляция фосфатурии и восстановление нормофосфатемии. FGF-23 уменьшает уровень кальцитриола, что способствует увеличению секреции ПТГ. Снижение уровня FGF-23 повышает уровень кальцитриола и белка Клото [20, 42, 43].
В результате умеренного снижения почечной функции уменьшается экскреция фосфора и повышается его уровень в крови, активизируя синтез и нарастание FGF-23. Увеличение секреции ПТГ способствует дальнейшему нарастанию концентрации FGF-23 и развитию вторичного гиперпаратиреоза, коррелирующего с различными типами нарушений минерализации скелета у пациентов с ХБП (гиперпаратиреоидной остеодистрофией и фиброзным остеитом) [18, 43].
Данные исследований позволяют предположить, что лептин напрямую стимулирует образование FGF-23 в кости [44]. Синтез FGF-23 также связан с дислипидемией – повышением триглицеридов, дефицитом холестерина липопротеинов высокой плотности, гиперинсулинемией (от 8 до 12%), индексом HOMA (Homeostatic Model Assessment), высоким индексом массы тела, абдоминальным ожирением. Однако прямая корреляция FGF-23 с СД еще не доказана [45]. Несмотря на это, было продемонстрировано, что FGF-23 является независимым фактором риска ХПН у пациентов с диабетической нефропатией [46, 47].
Белок Клото*
Реализация эффектов FGF-23 на органы-мишени осуществляется белком Клото – бета-глюкозидазой, трансформирующей канонические рецепторы FGF-23 в специфические [48]. M. Kuro-o, открывший белок Клото, подчеркивал, что белок Клото будет занимать важное место в терапии таких заболеваний, как диабет, ожирение и ХБП.
В почках Клото опосредует экскрецию фосфатов и ингибирование синтеза кальцитриола. Уровень белка Клото уменьшается уже при начальном снижении функции почек и предшествует нарастанию концентраций Р и FGF-23, что может быть ранним признаком и инициатором минерально-костных нарушений у пациентов с ХБП. Установлено, что у пациентов с ХБП снижение экcпрессии матричной РНК белка Клото в почечной паренхиме связано с резистентностью органов-мишеней к действию FGF-23 и по механизму обратной отрицательной связи приводит к повышению уровня последнего.
У больных с терминальной стадией ХПН уровень FGF-23 может увеличиваться в 1000 раз по сравнению с нормой [20]. Несмотря на такое значительное повышение уровня FGF-23, оно не позволяет нормализовать баланс фосфора и приводит к снижению продукции кальцитриола. Это вызывает уменьшение всасывания кальция в кишечнике, гипокальциурию и умеренную гипокальциемию у пациентов с ХБП II–III стадии, ведет к развитию вторичного гиперпаратиреоза [49].
Считается, что применение фосфатсвязывающих препаратов для поддержания нормального уровня Р у пациентов с ХБП и дефицитом белка Клото может уменьшить или даже предотвратить минеральные и сосудистые расстройства [47, 50].
У мышей дефицит белка Клото вызывает гиперфосфатемию, ускоренное старение, развитие атеросклероза, кальциноза и остеопороза [47]. В последнее время получены данные о прямой корреляции уровня FGF-23 со степенью кальцификации сосудов [51] и гипертрофии левого желудочка [52]. Эффекты FGF-23 в норме и при патологии представлены в обзоре, проведенным Е.М. Шутовым (рис. 2) [49].
Кроме этого, белок Клото блокирует внутриклеточные химические сигналы, которые передаются посредством инсулина и инсулиноподобных факторов роста, обеспечивая в определенной мере чувствительность организма к инсулину. Опыты на мышах показали, что уровень инсулина в крови трансгенных мышей был гораздо выше, чем в контрольной группе, а у мышей, лишенных гена белка Клото, наоборот, значительно ниже нормального уровня [53].
Уровень белка Клото был впервые оценен с учетом различных значений гликированного гемоглобина (HbA1c). Образцы анализов были разделены на две группы: контрольная – с HbA1c < 6,5% и основная – с HbA1 ≥ 6,5% (общепринятый критерий диагностики сахарного диабета). В обеих группах уровень креатинина был в контрольном диапазоне. Значительное снижение уровня белка Клото было выявлено в основной группе с уровнем HbA1c ≥ 6,5% по сравнению с контрольными образцами крови (HbA1c < 6,5%; p < 0,001) [54]. Экспериментальные данные, полученные ранее на грызунах, позволили сделать вывод о том, что снижение уровня белка Клото может быть результатом диабетической нефропатии [55]. Возможно, что снижение уровня белка Клото в крови пациентов с СД связано также с диабетической нефропатией, но это предположение требует проведения дальнейших исследований.
Кальцификация сосудов
Сосудистая кальцификация у пациентов с минерально-костными нарушениями при ХБП представлена двумя основными формами – кальцификацией интимы сосудов в сочетании с атеросклеротическим поражением и кальцификацией медии сосудов (вариантом артериосклероза).
Артериальный медиакальциноз, или склероз Менкеберга, характеризуется отсутствием накопления липидов и клеток воспаления в сосудистой стенке и обусловлен фенотипической трансформацией гладкомышечных клеток в остеобластоподобные [56, 57]. Артериальный медиакальциноз превалирует у пациентов с минерально-костными нарушениями при ХБП и признан специфическим для пациентов с СД. Фенотипические и молекулярные признаки кальцификации медии у больных СД и пациентов с ХБП совпадают [58].
Патогенез эктопического окостенения, происходящего в гладких мышечных клетках, очень сложный, и нарушение минерального обмена играет ключевую роль в этом процессе. Повышение уровня фосфора >2,4 ммоль/л индуцирует кальцификацию гладкомышечных клеток in vitro. Подобно фосфору, повышение концентрации кальция (> 2,6 ммоль/л) в культуре медии приводит к минерализации и фенотипическому изменению гладкомышечных клеток в остеобластоподобные клетки [59]. В последнее время получены данные о прямой корреляционной связи уровня FGF-23 c кальцификацией сосудов, однако она еще не объяснена [49].
Между тем известно, что воспаление, уремия, концентрация ПТГ, Ca, P, остеонектина, остеопонтина и гомоцистеина играют важную роль в сосудистой кальцификации [15], которая является важным фактором риска сердечно-сосудистой заболеваемости и смертности пациентов с ХБП [2, 3, 12, 26, 27, 29, 60].
Заключение
Минеральные и костные нарушения могут выявляться при снижении СКФ менее 70 мл/мин/1,73м² еще до установления III стадии ХБП. Ухудшение почечной функции и прогрессирование минерально-костных нарушений при ХБП осложняются развитием вторичного гиперпаратиреоза. Пациенты с СД и ХБП подвержены риску развития более ранних и тяжелых нарушений минерально-костного обмена, которые являются ведущей причиной сердечно-сосудистой заболеваемости и смертности в этой группе больных. В этой связи необходимо внедрение новых методов диагностики ранних минерально-костных нарушений у пациентов с ХБП и СД и проведение терапии (фосфатсвязывающие препараты, витамин D), направленной на снижение риска сердечно-сосудистой заболеваемости и смертности.