Спинальная мышечная атрофия: клинические проявления и подходы к терапии
- Аннотация
- Статья
- Ссылки
- English
![Рис. 1. Множественность системных проявлений у взрослых со СМА [14]](/upload/resize_cache/iblock/a3c/d6qqd0d774km6sibyls91r0tnpwcb8jm/800_800_1/Chernysheva2.jpg)
![Рис. 2. Время дебюта моторных и немоторных проявлений у пациентов со СМА (0 – дебют моторных симптомов, А – СМА 1–2-го типов, Б – 3-го типа, В – 4-го типа) [14]](/upload/resize_cache/iblock/03f/2tyfumct9a7v4c8xucu4zp2elzvyngm7/800_800_1/Chernysheva3.jpg)
Спинальная мышечная атрофия (СМА) – редкое нервно-мышечное заболевание, развивающееся вследствие изменения строения и снижения уровня неизмененного белка SMN, ответственного за выживание двигательных альфа-мотонейронов передних рогов спинного мозга и моторных нейронов ствола головного мозга. Заболевание развивается в условиях биаллельных мутаций SMN1. Дефицит белка SMN приводит к прогрессирующей дегенерации мотонейронов с возникновением двигательных симптомов, таких как мышечная слабость и амиотрофия, в большей степени выраженных в проксимальных отделах конечностей и аксиальной мускулатуре, с последующим распространением периферического пареза на дыхательные мышцы, а также мышцы лица и глотки. В отсутствие лечения заболевания наступает летальный исход вследствие дыхательной недостаточности. До появления специфических методов лечения, изменяющих течение заболевания, СМА была вторым наиболее распространенным фатальным аутосомно-рецессивным заболеванием после муковисцидоза и наиболее распространенной генетической причиной детской смертности [1].
Предполагаемая частота СМА – один случай на 6–10 тыс. новорожденных [2]. Данные о распространенности заболевания в РФ отсутствуют [3], но, вероятно, соответствуют общемировым.
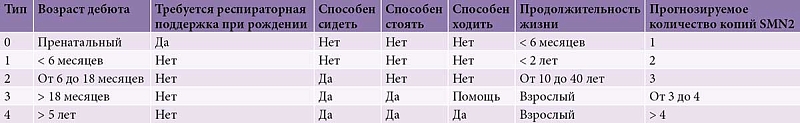
СМА подразделяется на несколько фенотипов (в настоящее время выделяют пять типов заболевания – 0, 1, 2, 3 и 4 (таблица)) в зависимости от времени развития. При этом тип 0 предполагает наличие двигательных симптомов уже при рождении, а тип 4 – дебют заболевания во взрослом возрасте. Возраст развития первых клинических симптомов, а также тяжесть заболевания обратно пропорциональны количеству белка SMN [4].
Самый распространенный вариант из аутосомно-рецессивных проксимальных СМА – СМА5q – обусловлен мутацией в гене SMN1 на длинном плече 5-й хромосомы. Наиболее распространенные формы СМА вызваны делециями экзонов 7 или 7–8, которые выявляются у 95% пациентов. Остальные 5% пациентов считаются компаунд-гетерозиготами по делеции в одной копии гена SMN1 и точковой мутации в другой, крайне редко – компаунд-гетерозиготами по двум минорным мутациям [5].
У человека в отличие от ряда других млекопитающих развитие заболевания обусловлено не только нарушением строения гена SMN1, но также изменением строения гена SMN2 – паралога (гена, возникшего в результате удвоения гена в пределах одного генома, однако не полностью аналогичного своей копии; такой ген может эволюционировать, приобретая новое значение и роль) гена SMN1. В 90% случаев транскрипции SMN2 происходит сплайсинг экзона 7, в результате чего образуется нестабильный белок SMN∆7. В то же время в оставшихся 10% образуется нормальный и функциональный белок SMN, способствующий выживанию спинномозговых двигательных нейронов и влияющий на тяжесть клинических проявлений [6]. Гены SMN1 и SMN2 более чем на 99% идентичны и расположены на хромосоме 5q13.2. Основное различие между ними заключается в замене цитозина на тимин в экзоне 7 SMN2. Потеря белка SMN1 частично компенсируется синтезом белка SMN2 – механизмом, который объясняет некоторую, но не всю фенотипическую изменчивость у пациентов со СМА [7]. Тяжесть заболевания при СМА, как правило, обратно коррелирует с числом копий SMN2, которое варьируется от 0 до 8 в нормальной популяции, и в меньшей степени – с уровнем белка SMN. Наличие четырех или более копий SMN2 связано с более мягким фенотипом [8]. Существует также ряд редких спинальных мышечных атрофий, не относящихся к 5q. СМА, не относящиеся к 5q, генетически и клинически гетерогенны [9].
Традиционно считалось, что сниженный уровень SMN вызывает избирательную гибель периферических мотонейронов, что приводит к развитию периферического пареза и атрофии скелетных мышц. Тем не менее многочисленные исследования клинических проявлений СМА продемонстрировали, что СМА на самом деле – мультисистемное расстройство. Появляется все больше данных, свидетельствующих о том, что SMN играет более значимую роль в функционировании различных типов клеток, органов и систем. Так, согласно результатам проведенных исследований, гены SMN1 и SMN2 экспрессируются в каждом типе тканей тела человека и, по сути, необходимы для поддержания жизнеспособности всех эукариотических клеток. Белок SMN1 принимает участие в следующих процессах:
- эндоцитоз и аутофагия, стабилизация цитоскелета: дефицит SMN приводит к снижению клеточного транспорта (например, синаптических пузырьков, гранул РНК и митохондрий) и эндоцитоза. При отсутствии SMN наблюдается не только дестабилизация микротрубочек, но и деполимеризация актинового цитоскелета, связанная с активацией пути RhoA – ROCK [10];
- процессинг/сплайсинг РНК: белок SMN присутствует как в ядре, так и в цитоплазме в составе SMN-комплексов, которые представляют собой самособирающиеся многомерные белковые структуры, необходимые для сплайсинга пре-мРНК. Белок SMN выступает в роли субъединицы SMN-комплекса при биосинтезе малых ядерных рибонуклеопротеиновых частиц (мяРНП; small nuclear ribonucleoprotein particles). SMN-комплекс позволяет ядерным Sm-белкам и обогащенным уридином малым ядерным РНК формировать мяРНП, участвующие в сплайсинге пре-мРНК различных генов;
- апоптоз: белок SMN модулирует апоптоз, блокируя активацию нескольких каспаз и других ключевых регуляторов выживания клетки [11];
- трансляции некоторых белков путем взаимодействия с рибосомами. а также репарация ДНК. Так, белок SMN увеличивает ассоциацию RAD51-ssDNA с гетерологичной двухцепочечной ДНК in vitro, поэтому предполагается, что он функционирует на этой стадии. Как только гомологичная область двухцепочечной ДНК идентифицирована, в нее вторгается одноцепочечная ДНК и происходит удлинение цепи. Для краткости описан простейший результат – отжиг цепи, зависящий от синтеза, при котором одна цепь удлиняется, а затем повторно отжигается с комплементарной цепью [12];
- ремоделирование РНК и других РНК-содержащих комплексов: SMN участвует в сборке и ремоделировании различных РНК-протеиновых комплексов, что необходимо для нормальной обработки РНК и транспорта внутри клетки;
- клеточная выживаемость и стресс-ответ: SMN также участвует в регуляции различных путей клеточной выживаемости и ответа на стресс, включая активацию протективных путей, что помогает клеткам справляться с повреждениями;
- дыхательная функция митохондрий: недостаточность белка SMN в клетке приводит к дефициту аденозинтрифосфата и, соответственно, повышению активности цитохром-c-оксидазы, а также увеличению потенциала мембраны митохондрии и, как следствие, окислительному стрессу. Изменения происходят не только в двигательных нейронах.
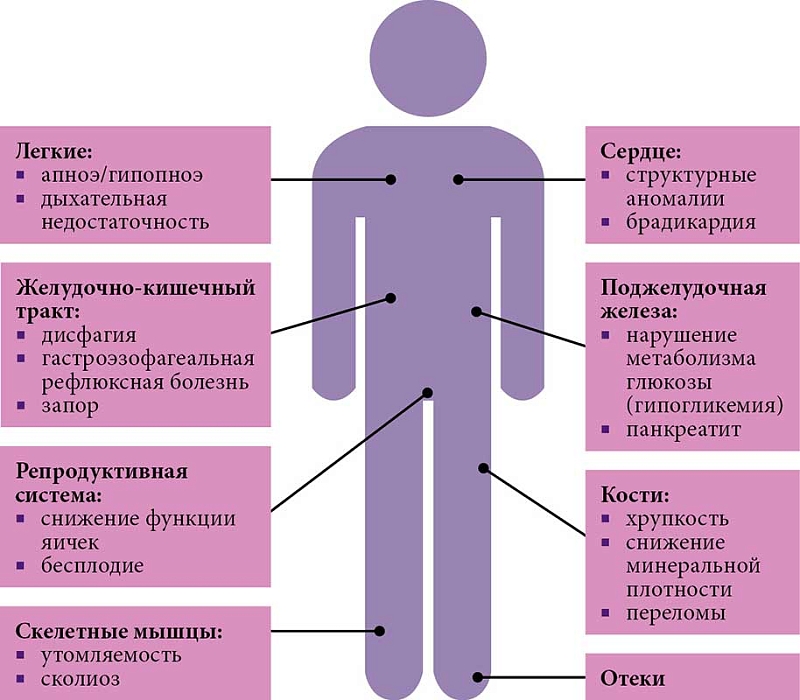
Таким образом, белок SMN поддерживает жизнедеятельность клетки на информационном и структурном уровне и является соединением, без которого жизнь любой ткани человеческого тела невозможна. С учетом повсеместной экспрессии белка SMN его дефицит помимо преобладающих неврологических нарушений обусловливает также и ряд системных нарушений. Это подтверждается как на экспериментальных моделях, так и в эпидемиологических исследованиях. G. Hamilton и T.H. Gillingwater выявили ряд клеток и тканей, являющихся патологическими мишенями за пределами традиционно изучаемой при СМА нервно-мышечной системы [13]. В опубликованной в 2019 г. обезличенной базе страховых претензий (США) было проанализировано 63 444 784 клинических случая страховых пациентов за период с 1 января 2008 г. по 1 октября 2015 г. За это время 1038 пациентов со СМА обращались к врачам по поводу немоторных жалоб. Были проведены два анализа. Первый касался претензий за весь период действия страхового покрытия, второй (в отношении пациентов со СМА) – претензий до постановки диагноза какого-либо нервно-мышечного заболевания или наличия признаков серьезной нервно-мышечной дегенерации. Проведенное исследование показало, что до появления первых клинических признаков нервно-мышечной дегенерации у пациентов со СМА отмечались нарушения сердечно-сосудистой, желудочно-кишечной, метаболической, репродуктивной и скелетной систем. Кроме того, заболевание обычно прогрессировало поэтапно, затрагивая все больше органов и систем до того, как развивались периферический парез и амиотрофия (рис. 1 и 2) [14].
В 2019 г. R. Günther и соавт. провели скрининг нейролептических симптомов (НЛС) у 70 взрослых пациентов со СМА 2-го и 3-го типов и 59 здоровых лиц контрольной группы, соответствовавших по половозрастным характеристикам пациентам основной группы, в рамках многоцентрового поперечного исследования, включавшего пять различных центров, специализирующихся на оказании медицинской помощи при заболеваниях мотонейронов [15]. Был использован опросник самооценки из 30 пунктов, касающихся желудочно-кишечных, вегетативных, нейропсихиатрических расстройств и нарушений сна, – опросник НЛС (NMS-Quest), который является валидированным инструментом при болезни Паркинсона. Общая тяжесть НЛС была низкой у взрослых пациентов со СМА и существенно не отличалась от таковой в контрольной группе. Общая частота НЛС у пациентов со СМА не коррелировала с показателями тяжести заболевания. Однако пункты «трудности с глотанием», «падения» и особенно «отеки ног» значительно чаще отмечались у пациентов со СМА. Частота встречаемости нейропсихиатрических симптомов была сопоставима с таковой в контрольной группе.
К наиболее частым немоторным симптомам относятся гиповентиляция и апноэ (рис. 3). Следует отметить, что гиповентиляция в значительной степени определяется слабостью межреберных мышц. В то же время апноэ, вероятнее всего, является центральным симптомом, связанным с поражением вегетативных структур ствола головного мозга [16]. Вовлеченность вегетативной нервной системы подтверждается в том числе частым развитием у пациентов со СМА ортостатической гипотензии и аритмии, преимущественно обусловленной слабостью влияний блуждающего нерва на сердце (эктопические желудочковые и суправентрикулярные аритмии). Часто встречаются запор, гастропарез, панкреатит. У пациентов со СМА нередко диагностируют гастроэзофагеальную рефлюксную болезнь. При этом она может отмечаться до появления слабости диафрагмы и других дыхательных мышц и усугубляться с их развитием. Типично грубое нарушение опорно-двигательного аппарата. Так, наряду с мышечной слабостью сколиоз и контрактуры суставов являются частыми осложнениями у пациентов со СМА, что ограничивает подвижность и приводит к болевому синдрому. В результате сниженной физической активности, увеличения числа и активности остеокластов, возможной недостаточности усвоения питательных веществ вследствие патологии желудочно-кишечного тракта может развиться остеопороз, увеличивающий риск переломов. У пациентов со СМА нередко имеют место нарушения эндокринной системы, включая нарушения метаболизма глюкозы (гипогликемия) из-за снижения числа В-клеток поджелудочной железы. Характерны задержка или недержание мочи из-за дисфункции мышц тазового дна. У мужчин со СМА часто развивается бесплодие вследствие нарушения сперматогенеза (изменение транскриптома и сплайсинга в сперматогенезе). Слабость респираторных мышц также повышает риск развития респираторных инфекций, в частности пневмонии, что является одной из основных причин летальных исходов у пациентов со СМА. Болезнь и ограничения физической активности нередко приводят к развитию тревожных и депрессивных состояний. Пациенты могут испытывать чувство социальной изоляции и беспомощности. Таким образом, множественность клинических проявлений СМА за пределами двигательных нарушений, несомненно, свидетельствует о мультисистемном характере заболевания.
В настоящее время для лечения СМА одобрены три орфанных препарата: нусинерсен (антисмысловой олигонуклеотид), онасемноген абепарвовек (генная терапия) и рисдиплам (модификатор сплайсинга пре-мРНК SMN2).
С учетом серьезности заболевания и острой необходимости в эффективном лечении эти препараты были разрешены в рамках ускоренной процедуры оценки, которая сокращает сроки рассмотрения заявки на регистрацию лекарственного средства Комитетом по лекарственным препаратам для человека Европейского агентства по лекарственным средствам (EMA) с 210 до 150 дней [17].
Нусинерсен стал первым орфанным препаратом для лечения СМА, одобренным Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов (FDA) в 2016 г., а затем и EMA в 2017 г. Нусинерсен относится к классу антисмысловых олигонуклеотидов и способен подавлять сплайсинг 7-го интрона гена SMN1, предотвращая связывание специфических репрессоров сплайсинга, hnRNPA1 и hnRNPA2, с ISS-N1, что позволяет интегрировать экзон 7 в конечный транскрипт и синтезировать полноразмерный белок SMN. Необходимость подавления сплайсинга 7-го интрона гена SMN1 вначале была доказана на экспериментальных моделях. Показано, что в результате терапии достоверно увеличивается уровень белка SMN, а также в 25 раз возрастает продолжительность жизни экспериментальных моделей СМА [18]. Кроме того, достоверно улучшаются двигательные функции (включая количество альфа-мотонейронов, среднюю площадь мышечных волокон, вес сердца, толщину межжелудочковой перегородки, стенки левого желудочка и целостность нервно-мышечных соединений). Более того, фармакокинетический анализ, проведенный на мышиных моделях и нечеловеческих приматах, показал хорошую биодоступность соединения на уровне центральной нервной системы при интратекальном введении. Главным недостатком применения нусинерсена является его неспособность преодолевать гематоэнцефалический барьер (ГЭБ) и, как следствие, необходимость многократных люмбальных пункций для интратекального введения. Это значительно снижает приверженность лечению, особенно у пациентов с тяжелым сколиозом.
Онасемноген абепарвовек был одобрен в 2020 г. как первая и на данный момент единственная генная терапия СМА. Показаниями к назначению данного лечения были возраст до двух лет и биаллельные мутации в гене SMN1 с тремя или менее копиями гена SMN2. Доклинические исследования на мышах и нечеловеческих приматах продемонстрировали способность препарата проникать через ГЭБ после внутривенного введения. При использовании данного метода лечения значительно увеличивается продолжительность жизни (медиана 282 дня для мышей, получавших лечение, против 17,5 дня для мышей SMAΔ7, не получавших лечения) и улучшаются двигательные функции (особенно у животных, получавших лечение на ранних стадиях) [19].
Успех нусинерсена послужил мощным стимулом для разработки более доступных (в том числе в отношении пути введения) методов лечения СМА. В ходе исследований было синтезировано перорально вводимое производное кумарина, впоследствии модифицированное для преодоления потенциальных проблем мутагенности in vitro и фототоксичности, обычно связанных с этим классом препаратов. Дальнейшая оптимизация соединения улучшила селективность и сродство к мишени, что позволило снизить дозу, необходимую для достижения терапевтического эффекта. Результатом исследований стало создание препарата рисдиплам, который был последовательно одобрен FDA и EMA в качестве первого перорального препарата для лечения СМА в 2020 и 2021 гг. соответственно [20]. Пероральный способ введения рисдиплама значительно повышает его биодоступность по сравнению с нусинерсеном. Это позволяет предположить, что данный вид лечения потенциально способен воздействовать как на моторные, так и на немоторные симптомы СМА.
Эффективность нусинерсена доказана в 13-месячном двойном слепом клиническом исследовании фазы III ENDEAR с участием 121 пациента со СМА с ранним дебютом в возрасте до шести месяцев. Этим пациентам на момент получения первой дозы было менее семи месяцев (типы 0, 1) [21]. Пациенты были в соотношении 2:1 рандомизированы на две группы – интратекальной инъекции нусинерсена (основная группа) и плацебо (контрольная группа). Согласно результатам исследования, 51% пациентов основной группы и все пациенты контрольной группы не достигли этапа двигательного развития, запланированного в клиническом исследовании как точка оценки эффективности. Риск смерти или необходимости постоянной инвазивной вентиляции легких был достоверно выше в контрольной группе (0,53; p = 0,005). В дальнейшем исследование ENDEAR было досрочно прекращено из-за результатов промежуточного анализа и этических соображений в отношении пациентов контрольной группы.
Исследование эффективности онасемногена абепарвовека проводилось на основании протокола STR1VE-US, открытого клинического исследования фазы III, включавшего 22 симптоматических пациента в возрасте до шести месяцев с диагнозом СМА 1-го типа [22]. Период наблюдения составил 18 месяцев. Первичная конечная точка эффективности (приобретение и сохранение способности сидеть без поддержки более 30 секунд) была достигнута у 59% пациентов основной группы. Ни одному из пациентов контрольной группы этой конечной точки достичь не удалось.
Эффективность рисдиплама оценивали в нескольких клинических исследованиях. Так, потенциальную эффективность у пациентов со СМА в исследовании FIREFISH анализировали при раннем дебюте заболевания [23], в исследовании SUNFISH – при позднем [24]. В открытое исследование FIREFISH был включен 41 младенец в возрасте от одного до семи месяцев с генетически подтвержденной СМА 1-го типа. Первичной конечной точкой служила способность сидеть без поддержки в течение не менее пяти секунд после 12 месяцев лечения – результат, который никогда не наблюдается у нелеченых пациентов со СМА 1-го типа. После 12 месяцев лечения 12 (29%) пациентов достигли указанной конечной точки. Таким образом, исследование показало, что рисдиплам эффективен при наиболее агрессивном течении СМА.
В рандомизированном двойном слепом плацебо-контролируемом клиническом исследовании фазы III SUNFISH участвовали пациенты в возрасте от двух до 25 лет с подтвержденным диагнозом СМА 2-го или 3-го типа. Пациенты были в соотношении 2:1 рандомизированы на две группы: 120 пациентов основной группы ежедневно принимали перорально рисдиплам, 60 пациентов контрольной группы – плацебо. Срок наблюдения оставил 12 месяцев. В исследовании отмечалось достоверное улучшение двигательной функции, причем наибольшим оно было у пациентов более младшего возраста (2–5 лет).
Как видно из описания дорегистрационных клинических исследований препаратов, одобренных для лечения СМА, все исследования, кроме SUNFISH, были открытыми, краткосрочными и включали небольшое количество пациентов. Такой тип клинических исследований в целом характерен для орфанных, в том числе быстро прогрессирующих, заболеваний, когда первичная оценка эффективности на небольших группах обычно достаточна для одобрения лекарственного препарата. В дальнейшем было проведено около 70 пострегистрационных клинических исследований [25].
Из проведенных исследований следует отметить:
- два открытых расширенных исследования SHINE и NURTURE, показавших, что прием повторных доз нусинерсена, вводимых интратекально, безопасен у младенцев с генетически диагностированной и досимптоматической СМА [26];
- несколько клинических исследований, продемонстрировавших безопасность применения онасемногена абепарвовека различными клиническими группами;
- четыре открытых расширенных исследования (FIREFISH, SUNFISH, JEWELFISH и RAINBOWFISH);
- два наблюдательных исследования, направленных на сбор данных о некоторых исходах беременности и осложнениях у женщин со СМА, получавших рисдиплам (исследование BN42833), а также на оценку влияния однократных пероральных доз рисдиплама на интервал QT электрокардиограммы (исследование BP42817) [25].
Накопление данных в результате клинической практики и назначения лекарственных препаратов позволило расширить знания об эффективности и безопасности патогенетической терапии СМА. Так, результаты проспективного трехлетнего исследования SMArtCARE показали стабилизацию прогрессирования заболевания у 231 амбулаторного пациента (114 детей (средний возраст – 8,6 года) и 117 взрослых (средний возраст – 37 лет)) одновременно с улучшением двигательной функции [27]. M. Pane и соавт. наблюдали умеренное улучшение двигательной активности и достоверное снижение смертности у детей, получавших терапию нусинерсеном [28]. Хорошие результаты получены в отношении эффективности и безопасности терапии онасемногеном абепарвовеком. Так, компания-производитель сообщает, что однократная внутривенная инфузия, введенная пациентам с симптоматической СМА, обеспечивает сохранение ранее достигнутых двигательных этапов развития даже спустя 7,5 года [29].
Таким образом, можно констатировать, что наши знания о природе, форме и клинических проявлениях СМА, значении генов SMN1 и SMN2 расширились в течение последних нескольких лет, а появление патогенетической терапии позволило существенно увеличить продолжительность и качество жизни пациентов. В настоящее время многие пациенты со СМА 1-го и 2-го типов перешагнули 18-летний рубеж, что было абсолютно невозможно еще несколько десятилетий назад. Вместе с тем эффективность существующей патогенетической терапии СМА нельзя считать абсолютной: излечение заболевания на фоне терапии не достигается, возможно развитие симптомов, ухудшающих качество жизни пациентов и приводящих к летальному исходу. Увеличение продолжительности жизни пациентов послужило основанием для гипотезы о мультисистемной природе СМА, выходящей за пределы нервной системы, что также определяет необходимость дальнейших исследований. Перспективы создания более эффективных методов лечения в ближайшее время связаны с углублением понимания патогенеза СМА.
E.S. Chernysheva, I.S. Preobrazhenskaya, PhD, Prof.
I.M. Sechenov First Moscow State Medical University
Contact person: Irina S. Preobrazhenskaya, preobrazhenskaya_i_s@staff.sechenov.ru
This article examines the main clinical manifestations of spinal muscular atrophy, including motor and non-motor symptoms. Data on the multisystem nature of spinal muscular atrophy are analyzed. The latest data on treatment options for this disease are presented, including the results of clinical trials evaluating the efficacy and safety of nusinersen, onasemnogene abeparvovec, and risdiplam.
Уважаемый посетитель uMEDp!
Уведомляем Вас о том, что здесь содержится информация, предназначенная исключительно для специалистов здравоохранения.
Если Вы не являетесь специалистом здравоохранения, администрация не несет ответственности за возможные отрицательные последствия, возникшие в результате самостоятельного использования Вами информации с портала без предварительной консультации с врачом.
Нажимая на кнопку «Войти», Вы подтверждаете, что являетесь врачом или студентом медицинского вуза.