Фиксированная доза пролголимаба 250 мг один раз в три недели для пациентов с метастатической меланомой. Результаты клинического исследования фазы III FLAT
- Аннотация
- Статья
- Ссылки
- English
Материал и методы. Многоцентровое несравнительное открытое исследование фазы III эффективности, фармакокинетики и безопасности было проведено с целью оценки сопоставимости режима дозирования пролголимаба 1 мг/кг один раз в две недели и фиксированной дозы 250 мг один раз в три недели у пациентов, ранее не получавших лечения по поводу неоперабельной или метастатической меланомы (BCD-100-8/FLAT, NCT05783882).
Основная цель исследования – доказательство неменьшей эффективности применения пролголимаба в дозе 250 мг один раз в три недели по сравнению с пролголимабом в дозе 1 мг/кг один раз в две недели для лечения пациентов с нерезектабельной или метастатической меланомой по показателю частоты объективного ответа (ЧОО) согласно критериям RECIST 1.1. Пациенты из клинического исследования MIRACULUM (BCD-100-2/MIRACULUM, NCT03269565) представляли собой ретроспективную контрольную группу.
Результаты. 114 пациентов получали пролголимаб в дозе 250 мг один раз в три недели и 61 пациент получал пролголимаб (Пролго) в дозе 1 мг/кг один раз в две недели (ретроспективный контроль). ЧОО была достигнута у 33,3% (95% доверительный интервал (ДИ) 24,8–42,8) пациентов в группе Пролго 250 мг по сравнению с 32,8% (95% ДИ 21,3–46,0) пациентов в группе Пролго 1 мг/кг. Разность рисков составила 0,00 (95% ДИ -0,12 – не достигнута), p = 0,0082. Оба терапевтических режима обладали благоприятным и сопоставимым профилем безопасности. При применении фиксированной дозы 250 мг один раз в три недели были продемонстрированы более высокие значения фармакокинетических показателей по сравнению с режимом 1 мг/кг. Ни у одного из пациентов не было выявлено связывающих антител к пролголимабу.
Заключение. Выбранный режим дозирования пролголимаба с фиксированной дозой 250 мг один раз в три недели обладает показателями эффективности и безопасности, сопоставимыми с режимом дозирования 1 мг/кг один раз в две недели.
Материал и методы. Многоцентровое несравнительное открытое исследование фазы III эффективности, фармакокинетики и безопасности было проведено с целью оценки сопоставимости режима дозирования пролголимаба 1 мг/кг один раз в две недели и фиксированной дозы 250 мг один раз в три недели у пациентов, ранее не получавших лечения по поводу неоперабельной или метастатической меланомы (BCD-100-8/FLAT, NCT05783882).
Основная цель исследования – доказательство неменьшей эффективности применения пролголимаба в дозе 250 мг один раз в три недели по сравнению с пролголимабом в дозе 1 мг/кг один раз в две недели для лечения пациентов с нерезектабельной или метастатической меланомой по показателю частоты объективного ответа (ЧОО) согласно критериям RECIST 1.1. Пациенты из клинического исследования MIRACULUM (BCD-100-2/MIRACULUM, NCT03269565) представляли собой ретроспективную контрольную группу.
Результаты. 114 пациентов получали пролголимаб в дозе 250 мг один раз в три недели и 61 пациент получал пролголимаб (Пролго) в дозе 1 мг/кг один раз в две недели (ретроспективный контроль). ЧОО была достигнута у 33,3% (95% доверительный интервал (ДИ) 24,8–42,8) пациентов в группе Пролго 250 мг по сравнению с 32,8% (95% ДИ 21,3–46,0) пациентов в группе Пролго 1 мг/кг. Разность рисков составила 0,00 (95% ДИ -0,12 – не достигнута), p = 0,0082. Оба терапевтических режима обладали благоприятным и сопоставимым профилем безопасности. При применении фиксированной дозы 250 мг один раз в три недели были продемонстрированы более высокие значения фармакокинетических показателей по сравнению с режимом 1 мг/кг. Ни у одного из пациентов не было выявлено связывающих антител к пролголимабу.
Заключение. Выбранный режим дозирования пролголимаба с фиксированной дозой 250 мг один раз в три недели обладает показателями эффективности и безопасности, сопоставимыми с режимом дозирования 1 мг/кг один раз в две недели.


Введение
Разработка ингибиторов иммунных контрольных точек (ИИКТ) произвела настоящую революцию в способах лечения пациентов со злокачественными новообразованиями, обеспечив достижение глубоких и устойчивых ответов на лечение, что привело к значительному улучшению показателей выживаемости больных различных нозологических групп.
В настоящее время в клинической практике широко применяются ИИКТ PD-1 класса иммуноглобулинов изотипа G (IgG) (пролголимаб, ниволумаб, пембролизумаб, тислезумаб и др.). Результаты клинических исследований (КИ) ИИКТ, доступные на данный момент (за исключением ипилимумаба), не выявили дозолимитирующей токсичности или дозозависимой эффективности, при этом наблюдаются тенденции к применению доз, значительно превышающих минимальные эффективные [1]. Объем распределения этих препаратов близок к объему плазмы, при этом важную роль играют ограниченное распределение в тканях и аффинность связывания целевого антигена. Метаболизм и выведение не оказывают влияния на функцию почек и печени из-за высокой молекулярной массы и отсутствия вовлечения ферментов системы цитохрома 450. Метаболизм и выведение происходят как специфическим (мишень-опосредованным, быстрым), так и неспецифическим (FcRn-опосредованным, медленным) путями, что приводит к нелинейному и линейному выведению соответственно. Насыщение целевого пути происходит быстро, при этом неспецифический путь становится преобладающим, что объясняет длительный период полувыведения этих соединений и медленный клиренс. Помимо этого, в нескольких исследованиях описываются взаимосвязи «экспозиция – ответ», при которых только концентрации оказывается недостаточно, однако определение клиренса этих ИИКТ, включающее различные ковариаты, кажется перспективным [2–5]. Доступные на данный момент ингибиторы PD-1 представляют собой иммуноглобулины изотипа G (IgG), обладающие схожими фармакокинетическими свойствами: не имеют дозозависимого эффекта с традиционной точки зрения, так как насыщение рецепторов PD-1 происходит при низких концентрациях препарата [6, 7]. Это объясняется высокой аффинностью препарата к рецепторам PD-1. Следовательно, эффективность ингибиторов PD-1 не является зависящей от дозы, а управляется биологическими факторами и воздействием на иммунную систему. Однако доза может влиять на развитие нежелательных явлений (НЯ) и переносимость препарата, поэтому в ряде ситуаций может потребоваться подбор оптимальной дозы для каждого пациента.
В 2020 г. по результатам рандомизированного КИ фазы II/III MIRACULUM (NCT03269565) в Российской Федерации был одобрен первый российский оригинальный ингибитор PD-1 пролголимаб (Фортека®, АО «Биокад») для лечения метастатической или нерезектабельной меланомы. В настоящее время показания к применению пролголимаба расширяются. В конце 2023 г. на основании КИ фазы III DOMAJOR (NCT03912389) в Российской Федерации был одобрен пролголимаб в комбинации с платиносодержащей химиотерапией в качестве терапии первой линии у пациентов с распространенным немелкоклеточным раком легкого (НМРЛ) [8]. В настоящее время проводятся два КИ фазы III: исследование эффективности и безопасности применения пролголимаба в комбинации с химиотерапией на основе препаратов платины с бевацизумабом и без него у пациентов с распространенным раком шейки матки в качестве терапии первой линии (FERMATA, NCT03912415) и КИ применения пролголимаба после неоадъювантной терапии нурулимабом + пролголимабом у пациентов с резектабельной меланомой III стадии и по достижении полного или почти полного патоморфологического ответа, определяемого по индексным лимфатическим узлам (NEOMIMAJOR, NCT05751928).
Пролголимаб – это рекомбинантное моноклональное антитело IgG1 с модифицированным Fc-фрагментом. Наличие мутации LALA (Leu234Ala/Leu235Ala) в Fc-фрагменте пролголимаба (замена двух аминокислот лейцина на аланин) минимизирует эффекторные свойства антитела. Таким образом, пролголимаб не связывается с FcyR-рецепторами макрофагов, что усиливает защиту популяции активированных Т-лимфоцитов от возможного антителозависимого фагоцитоза макрофагами и тем самым усиливает противоопухолевый эффект. Уникальный эпитоп связывания пролголимаба обеспечивает высокую насыщенность PD-1-рецепторов при минимальной концентрации препарата. В фазе II исследования MIRACULUM объективный ответ был достигнут у 38,1% пациентов с метастатической меланомой, получавших пролголимаб в виде монотерапии, выживаемость без прогрессирования (ВБП) в течение 24 месяцев – у 33,3% и общая выживаемость (ОВ) в течение 24 месяцев – у 57,1% пациентов. НЯ всех степеней тяжести, связанные с лечением, были зарегистрированы у 55,6% пациентов, 3–4-й степени тяжести – у 12,7%, серьезные НЯ и НЯ, приведшие к отмене лечения, – у 3,2 и 3,2% пациентов соответственно. Эффективность пролголимаба сопоставима с другими препаратами класса ингибиторов PD-1, а профиль безопасности оказался при непрямом сравнении наиболее благоприятным [9, 10].
Изначально пролголимаб, как и другие анти-PD-1-препараты (ниволумаб, пембролизумаб, тислезумаб и т.д.), изучался и был одобрен для применения в дозах, зависящих от массы тела: 1 мг/кг один раз в две недели [9, 11] для пациентов с метастатической меланомой и 3 мг/кг один раз в три недели для пациентов с распространенным НМРЛ. Результаты клинического исследования фазы I продемонстрировали, что максимально переносимая доза пролголимаба не была достигнута и не было обнаружено дозозависимого ответа, что соответствует данным других ингибиторов PD-1 [12]. Принимая во внимание биологический механизм действия ИИКТ [1], специфические свойства моноклональных антител (избирательный механизм действия, значительно более широкий терапевтический индекс) [2, 13], преимущества фиксированной дозы (повышенное удобство, устранение неэффективного расходования, повышение уровня безопасности за счет уменьшения ошибок дозирования, более точное соблюдение режима дозирования) [3], накопленные данные о других анти-PD-1-препаратах [4, 14–17], результаты исследований связи экспозиции и эффективности, экспозиции и безопасности, а также фармакокинетики (ФК) пролголимаба в исследованиях ранних фаз [5, 9, 12], было проведено многоцентровое несравнительное открытое исследование эффективности, ФК и безопасности с целью доказательства неменьшей эффективности пролголимаба в дозе 250 мг один раз в три недели по сравнению с ретроспективными данными для пролголимаба в дозе 1 мг/кг один раз в две недели у пациентов с нерезектабельной или метастатической меланомой, а также для сбора данных по ФК и безопасности (BCD-100-8/FLAT, NCT05783882).
Материал и методы
Дизайн исследования и лечение
Исследование BCD-100-8/FLAT (NCT05783882) представляло собой многоцентровое открытое исследование эффективности, ФК и безопасности применения пролголимаба (Пролго) в фиксированной дозе (250 г один раз в три недели) у пациентов с нерезектабельной или метастатической меланомой.
Фиксированная доза Пролго один раз в три недели (Q3W) была выбрана на основе популяционного фармакокинетического моделирования, а также данных об эффективности и безопасности схем лечения на основе массы тела из исследования BCD-100-2/MIRACULUM (NCT03269565). В анализ ФК были включены пациенты, получавшие Пролго в дозе 1 мг/кг один раз в две недели (Q2W) (n = 63) и 3 мг/кг Q3W (n = 61) в основной (фаза II) части КИ MIRACULUM. Для моделирования ФК препарата применялась двухкомпартментная модель. Анализ популяционной ФК продемонстрировал, что при применении Пролго в дозе 250 мг Q3W предполагаемые значения Cmax, Cav и Ctrough после первой и десятой инфузий были сравнимы со значениями, наблюдаемыми для дозы 3 мг/кг Q3W, однако были выше, чем значения, наблюдаемые для дозы 1 мг/кг Q2W. Моделирование эффективности и результаты завершенного исследования MIRACULUM показали отсутствие зависимости «доза – ответ». На основании вышеизложенного ожидалось, что Пролго в режиме дозирования 250 мг Q3W будет столь же эффективным, как и одобренный режим дозирования 1 мг/кг Q2W.
В КИ FLAT участвовали пациенты в возрасте ≥ 18 лет с неоперабельной или метастатической меланомой, ранее не получавшие терапию по поводу нерезектабельного или метастатического заболевания. Основные критерии включения: наличие образца опухоли для иммуногистохимического исследования перед началом терапии; общая оценка по шкале ECOG 0 или 1; наличие минимум одного измеримого таргетного очага в соответствии с критериями RECIST 1.1, подтвержденное независимым центральным пересмотром; а также предоставленное информированное согласие пациента в письменном виде. Пациенты не могли быть включены, если они ранее получали какое-либо лечение по поводу нерезектабельной (или метастатической) меланомы, или терапию анти-CTLA4- и/или анти-PD-1/PD-L1/PD-L2-препаратами, или таргетную терапию.
Пациенты получали Пролго внутривенно в дозе 250 мг Q3W до развития непереносимой токсичности или прогрессирования заболевания (в зависимости от того, что произошло раньше).
Пациенты из КИ MIRACULUM составляли ретроспективную контрольную группу. Данные по эффективности и безопасности режима дозирования с фиксированной дозой (250 мг Q3W) сравнивались с ранее не опубликованными данными, полученными для режима дозирования на основе массы тела 1 мг/кг Q2W в подтверждающей части (фаза III) КИ MIRACULUM. Данные по ФК, полученные при фиксированном режиме дозирования, сравнивались с ранее не опубликованными данными, полученными при использовании двух режимов дозирования на основе массы тела в основной части (фаза II) КИ MIRACULUM (1 мг/кг Q2W и 3 мг/кг Q3W).
КИ FLAT проводилось в тех же условиях, что и подтверждающая часть КИ MIRACULUM. Критерии включения пациентов, исследовательские центры, процедуры оценки эффективности и безопасности, разрешенная предшествующая и сопутствующая терапия по поводу основного заболевания были идентичными в обоих исследованиях. В подтверждающую часть КИ MIRACULUM вошли все пациенты из основной части исследования, получавшие Пролго в дозе 1 мг/кг Q2W в качестве терапии первой линии, а также были включены 15 дополнительных пациентов.
Первичной конечной точкой была частота объективного ответа (ЧОО) в соответствии с критериями RECIST 1.1 в течение 25 недель лечения. Вторичные конечные точки включали в себя ЧОО в соответствии с критериями irRECIST, частоту достижения контроля над заболеванием (ЧКЗ), время до ответа на терапию (ВДО) и длительность ответа на терапию (ДО). ЧКЗ, ВДО и ДО оценивались согласно критериям RECIST 1.1 и irRECIST.
Конечными точками для безопасности были доля пациентов с НЯ любой степени тяжести, тяжелыми НЯ (степень ≥ 3), серьезными НЯ, НЯ любой степени тяжести, связанными с исследуемым препаратом, иммуноопосредованными НЯ (иоНЯ) любой степени тяжести, тяжелыми иоНЯ (степень ≥ 3), доля пациентов, которым была произведена отмена терапии по причине НЯ, а также доля пациентов, которым была произведена отмена терапии по причине иоНЯ. Тяжесть НЯ оценивалась в соответствии с критериями CTCAE, версия 4.03.
Показатели ФК включали Cav, AUC0-t, Cmax, AUC0-∞, T1/2, Kel, Cl, Ctrough после однократного применения; AUCτ,ss, Cav,ss, Cmax,ss, Ctrough,ss после многократного применения.
Протокол исследования и поправки к нему были рассмотрены комитетами по этике в каждом исследовательском центре. Исследование проводилось в соответствии с принципами Хельсинкской декларации и рекомендациями надлежащей клинической практики. Перед включением в исследование все пациенты предоставили информированное согласие в письменном виде.
Оценка
Оценку размера опухоли проводили с помощью компьютерной томографии/магнитно-резонансной томографии в ходе скрининга в 57-й, 113-й и 169-й дни исследования независимо от приостановки исследуемой терапии. Ответ опухоли на лечение оценивался в соответствии с критериями RECIST 1.1 и irRECIST, все конечные точки эффективности были основаны на оценках, выполненных независимым центральным наблюдательным комитетом в режиме работы с обезличенными данными.
В анализы для оценки безопасности входили оценка показателей жизненно важных функций, физикальное обследование, электрокардиограмма, эхокардиограмма, забор крови для биохимического анализа сыворотки крови, общего анализа крови, коагулограммы и анализа функции щитовидной железы, анализ мочи, а также оценка нежелательных явлений. НЯ классифицировались с использованием общих терминологических критериев CTCAE, версия 4.03.
График визитов и проведения процедур в КИ FLAT соответствовал графику в КИ MIRACULUM.
Статистический анализ
Первичным эстимандом в исследовании является средний популяционный эффект терапии BCD-100 в режиме 250 мг один раз в три недели, оцениваемый с помощью ЧОО за 25 недель лечения, относительно эффекта терапии BCD-100 в режиме 1 мг/кг один раз в две недели в предположении, что отсутствие данных об ответе по любой причине, а также прием запрещенной терапии являются признаком недостаточного эффекта терапии (такие субъекты расценены как неответчики). Таким образом, в настоящем исследовании была предусмотрена композитная стратегия учета следующих интеркуррентных событий для первичного эстиманда: выбывание из исследования по любой причине, прием запрещенной терапии.
В подтверждающей части КИ MIRACULUM наблюдалась ЧОО 20/58 (34,48%) в популяции PP (per protocol, пациенты, прошедшие как минимум одно плановое контрольное исследование компьютерной томографии) и 20/61 (32,79%) в популяции mITT.
Для доказательства гипотезы неменьшей эффективности требовалось, чтобы минимум 91 пациент продемонстрировал неменьшую эффективность с границей неменьшей эффективности -0,2 для нижнего предела доверительного интервала (ДИ) при разности рисков с 80%-ной мощностью и односторонним уровнем значимости 0,05 (с использованием частоты ответа для популяции PP из подтверждающей части исследования MIRACULUM как ожидаемой ЧОО при расчете размера выборки). Принимая во внимание возможный процент выбывания пациентов 17%, в исследование запланировано включить не менее 110 участников.
Первичный анализ проводился с использованием программы SAS PROC GENMOD. Односторонний стратифицированный 95%-ный ДИ правдоподобной вероятности для разницы рисков, определяемый как доля пациентов с объективным ответом между сравниваемыми схемами терапии, был получен с использованием оценки параметров терапии на основе модели, подходящей для альтернативной гипотезы. Значение p для неменьшей эффективности было рассчитано с использованием статистики отклонений, которая представляет собой удвоенную разницу в значениях логарифмического правдоподобия между двумя моделями. Отсутствующие по какой-либо причине данные для первичной конечной точки были учтены в анализе с использованием подстановки недостающих данных при отсутствии ответа.
Основной анализ эффективности и безопасности проводился для популяции mITT (пациенты, получившие хотя бы одну дозу препарата). В популяцию для оценки ФК после однократного введения вошли все получившие инфузию препарата пациенты, у которых отсутствовали не более трех доступных образцов крови после первого введения. Популяция для анализа ФК после многократного введения включала всех пациентов, у которых отсутствовали не более трех образцов крови после пятого введения препарата. Популяция для анализа Ctrough включала пациентов, которые получили хотя бы одно введение, у них имелись хотя бы одно доступное значение концентрации перед первым введением и хотя бы одно значение концентрации перед любым последующим введением. В популяцию для оценки иммуногенности вошли все включенные в исследование пациенты, получившие хотя бы одно введение исследуемого препарата, с доступными (поддающимися оценке) образцами сыворотки крови, взятыми до введения исследуемого препарата и хотя бы при одном последующем визите.
Статистический анализ проводился с помощью SAS версии 9.4 (SAS Institute Inc, Кэри, Северная Каролина).
Результаты
Исследуемая популяция
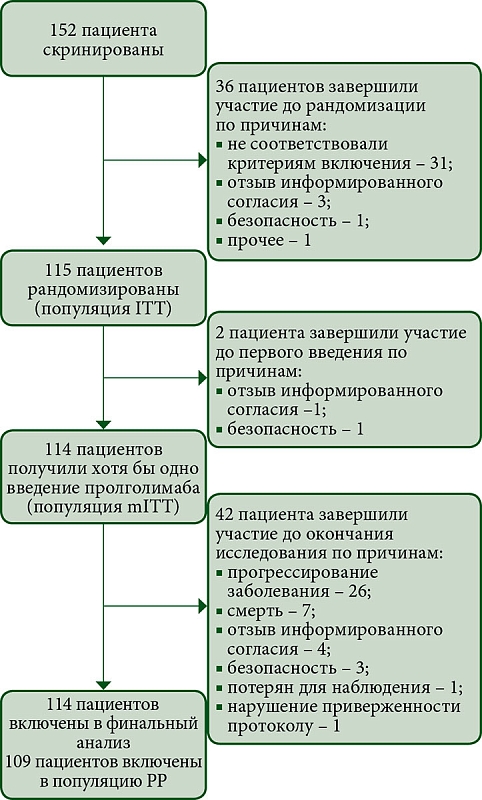
КИ FLAT было начато 13 мая 2022 г., 114 пациентов из 14 центров РФ получали Пролго 250 мг Q3W (группа Пролго 250 мг, рис. 1). 61 пациент из КИ MIRACULUM, получавший Пролго 1 мг/кг Q2W, составил контрольную группу (популяция mITT в фазе III, группа Пролго 1 мг/кг).
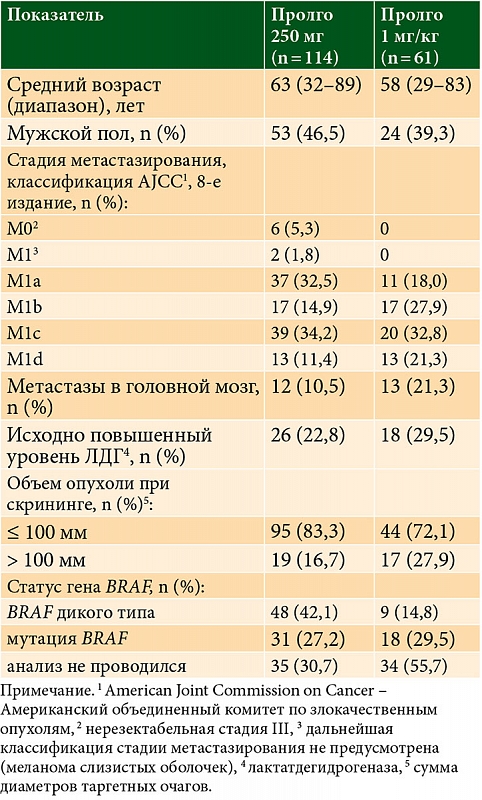
Исходные демографические и клинические характеристики пациентов в целом были сбалансированы между группами (табл. 1). Все пациенты в обеих группах получали Пролго в качестве терапии первой линии. Количество пациентов с меланомой некожных локализаций (меланомой слизистых оболочек или увеальной меланомой) в группе Пролго 250 мг – 4/114 (3,5%), в группе Пролго 1 мг/кг – 1/61 (1,6%). Медиана наблюдения – 5,6 месяца в группе Пролго 250 мг, 4,6 месяца в группе Пролго 1 мг/кг для анализа безопасности и 12,9 месяца для анализа эффективности.
Эффективность
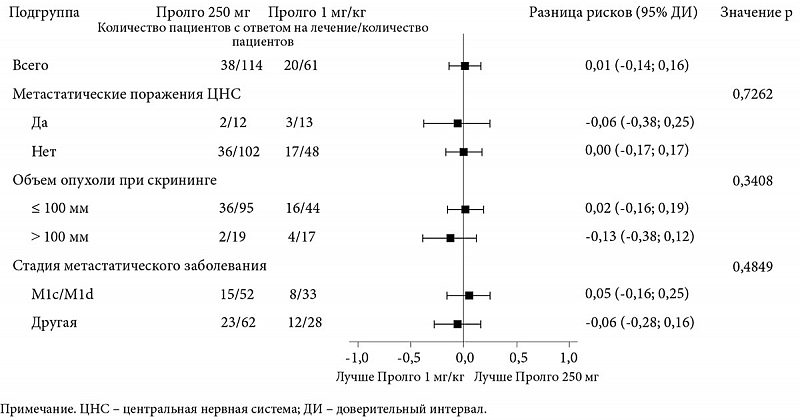
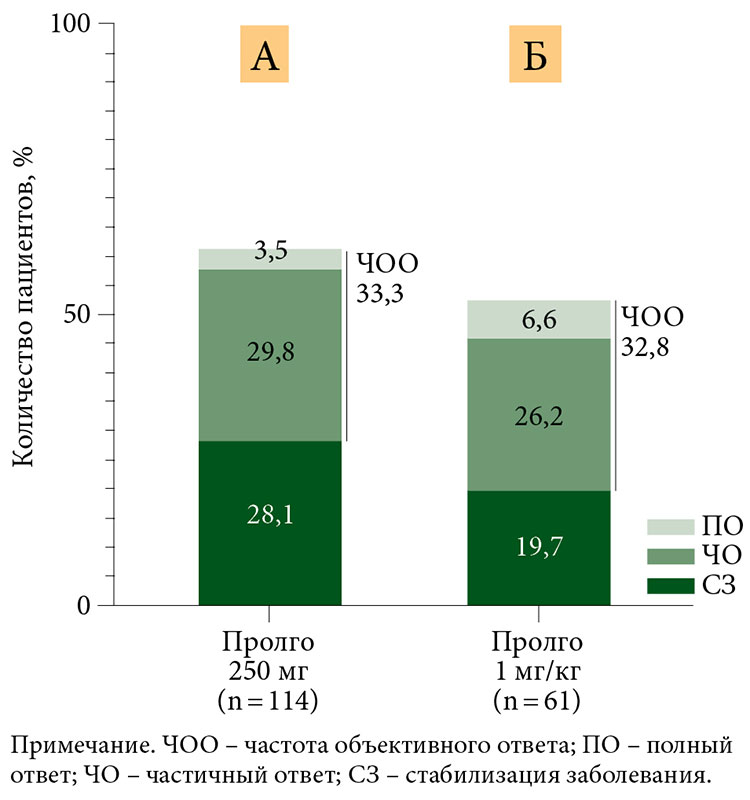
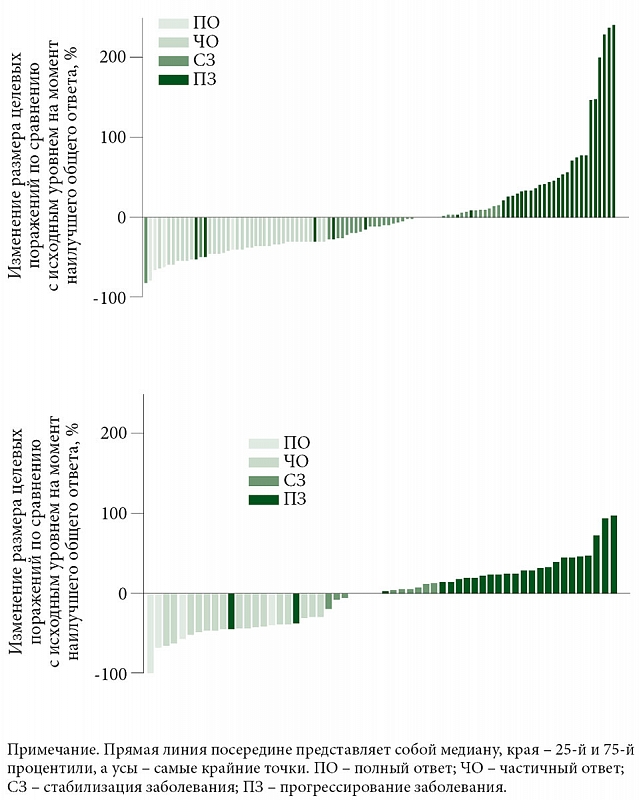
По данным независимой централизованной оценки, у 38 из 114 (33,3%) пациентов в группе Пролго 250 мг наблюдался ответ на лечение в соответствии с критериями RECIST 1.1 по сравнению с 20 из 61 (32,8%) в группе Пролго 1 мг/кг (разница рисков (РР) 0,00, 95% ДИ 0,12 – не достигнута (НД), p = 0,0082) (табл. 2), что отвечало критерию неменьшей эффективности. Результаты для ЧОО были одинаковыми в заранее определенных подгруппах (рис. 2).
Среднее время достижения объективного ответа в группе Пролго 250 мг (РР 1,906, 95% ДИ 1,873–2,103) и группе Пролго 1 мг/кг (РР 2,070, 95% ДИ 1,873–3,680) было одинаковым. Медиана продолжительности ответа на терапию не была достигнута в группе Пролго 250 мг (95% ДИ 3,745–НД) и составила 9,429 месяца (95% ДИ 4,862–НД) для группы Пролго 1 мг/кг. Глубина ответа на лечение определялась как изменение размера таргетных очагов по сравнению с исходным уровнем на момент наилучшего общего ответа (рис. 3).
Ответ на терапию в соответствии с критериями irRECIST был зарегистрирован у 41 из 114 (36,0%) пациентов в группе Пролго 250 мг и у 26 из 61 (42,6%) пациентов в группе Пролго 1 мг/кг (табл. 2).
Фармакокинетика и иммуногенность
В настоящем анализе фармакокинетические данные, полученные при режиме дозирования с фиксированной дозой, сравнивались с ранее не опубликованными данными, полученными при использовании двух режимов дозирования на основе массы тела в КИ MIRACULUM (1 мг/кг Q2W и 3 мг/кг Q3W).
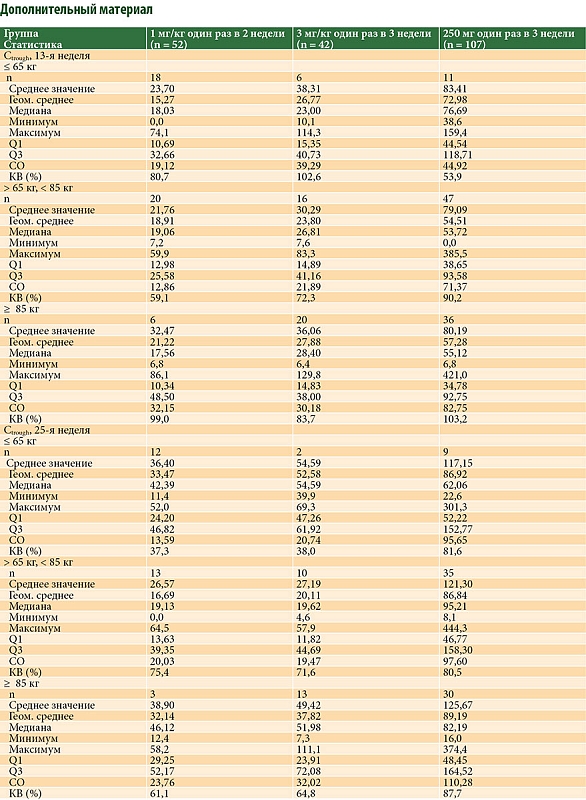
На рис. 4 показано, что в общей популяции для оценки Ctrough значения Ctrough при применении фиксированной дозы были стабильно выше, чем при режиме дозирования на основе массы тела. При фиксированной дозе определялась адекватная экспозиция для всех подгрупп по массе тела, так как максимальное и минимальное средние геометрические значения Ctrough для Пролго в дозе 250 мг были выше, чем для Пролго в дозе 1 мг/кг Q2W и 3 мг/кг Q3W (дополнительный материал, табл. S1).
Ни у одного из пациентов, вошедших в популяцию оценки иммуногенности (n = 95), не было выявлено связывающих антител к пролголимабу.
Безопасность
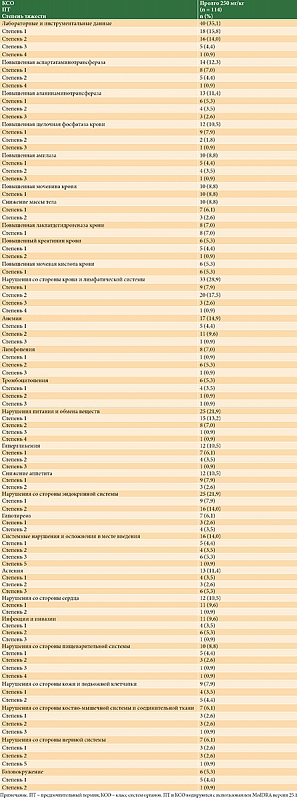
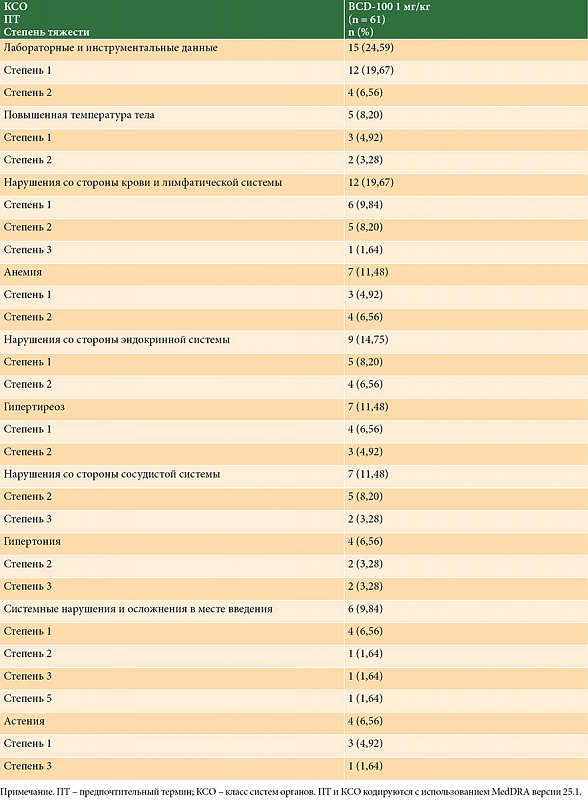
Режим дозирования Пролго с фиксированной дозой имел благоприятный профиль безопасности: не было зафиксировано ни одного связанного с терапией летального исхода. НЯ любой степени тяжести были зарегистрированы у 85/114 (74,6%) пациентов, у 21 из 114 (18,4%) пациентов наблюдалось минимум одно НЯ ≥ 3-й степени, у 60 из 114 пациентов (52,6%) наблюдалось минимум одно НЯ, связанное с исследуемой терапией. Профили безопасности для режимов Пролго 250 мг Q3W и Пролго 1 мг/кг Q2W были сопоставимы (табл. 3). НЯ, связанные с терапией, возникали с одинаковой частотой при применении фиксированной дозы и при режиме 1 мг/кг, однако частота иоНЯ была ниже в группе Пролго 250 мг. Прекращение терапии по причине НЯ потребовалось двум пациентам из 114 (1,8%) в группе Пролго 250 мг и трем пациентам из 61 (4,9%) в группе Пролго 1 мг/кг. Частота и профиль НЯ были сопоставимыми в обеих группах (дополнительный материал, табл. S2 и S3). Наиболее частые НЯ (> 5% пациентов в обеих группах) указаны в приложении.
Обсуждение
В настоящее исследование были включены пациенты с нерезектабельной или метастатической меланомой, которые ранее не получали терапию по поводу данного заболевания, а также терапию анти-CTLA4- и/или анти-PD-1/PD-L1/PDL-2-препаратами или таргетную терапию. Популяция пациентов была подобрана так, что она полностью соответствовала третьему этапу исследования BCD-100-2/MIRACULUM (ретроспективный контроль).
Использование ретроспективного контроля для демонстрации неменьшей эффективности обусловлено возможностью объективной оценки и контроля показателей, влияющих на количество пациентов с ответом на лечение, которые не зависят непосредственно от эффективности препарата. Такими факторами являются популяция пациентов, определяемая критериями включения, предшествующая и сопутствующая терапия, методы и сроки оценки эффективности, место проведения исследования, а также такие показатели, как исходный размер опухоли, общее состояние пациента, сопутствующие заболевания и наличие метастазов в центральную нервную систему.
Первичной конечной точкой исследования является ЧОО, определяемая как полный или частичный ответ в соответствии с критериями RECIST 1.1 на 25-й неделе лечения согласно оценке независимого центрального пересмотра. Для данного исследования была выбрана эта конечная точка в силу объективности оценки эффективности противоопухолевых препаратов. ЧОО является наиболее чувствительным параметром, который используется в исследованиях эффективности противоопухолевых препаратов. В отличие от других стандартных параметров для клинических исследований в онкологии, таких как ВБП и ОВ, ЧОО характеризует прямое воздействие препарата на опухоль. В качестве вторичных конечных точек эффективности, включающих ЧОО в соответствии с критериями irRECIST, анализировались параметры выживаемости. Факторы риска, влияющие на развитие объективного ответа на лечение пролголимабом у пациентов с нерезектабельной или метастатической меланомой, могут быть определены по результатам уже завершившегося клинического исследования MIRACULUM и учтены при статистическом анализе данных для сравнения эффективности лечения по результатам настоящего исследования с эффективностью лечения в КИ MIRACULUM. Таким образом, хотя в КИ FLAT применялся ретроспективный контроль, доступность результатов КИ MIRACULUM в сочетании с объективностью оценки первичной конечной точки позволяет полноценно сравнить эффективность двух схем лечения пролголимабом: 250 мг Q3W по сравнению с 1 мг/кг Q2W.
С этой же целью КИ FLAT проводилось только в тех исследовательских центрах, которые участвовали в КИ MIRACULUM. Критерии отбора в КИ FLAT были такими же, как и в исследовании MIRACULUM. Таким образом, результаты исследования BCD-100-2/MIRACULUM можно было использовать в качестве ретроспективного контроля, чтобы продемонстрировать неменьшую эффективность применения пролголимаба в дозе 250 мг Q3W по сравнению с дозой 1 мг/кг Q2W с точки зрения общего ответа на лечение.
Популяция III фазы в КИ MIRACULUM включала 1/61 (1,6%) пациента с меланомой некожных локализаций (меланома слизистых оболочек или увеальная меланома). Пациенты с меланомой некожных локализаций менее чувствительны к анти-PD-1-препаратам, таким как пролголимаб. В связи с этим и для обеспечения адекватного сравнения режимов дозирования пролголимаба 1 мг/кг Q2W и 250 мг Q3W в исследовании BCD-100-8/FLAT набор пациентов с меланомой некожных локализаций был ограничен с целью обеспечения сопоставимого количества таких пациентов: 4/114 пациентов (3,5%).
На основании вышеизложенного можно сделать вывод о том, что дизайн КИ FLAT дает возможность объективно сравнить эффективность двух схем лечения пролголимабом.
Данные о ФК препарата, представленные в этой статье, демонстрируют несколько более высокие значения фармакокинетических параметров при применении пролголимаба в фиксированной дозе 250 мг Q3W по сравнению с дозой 1 мг/кг один раз в две недели (первоначально одобренная доза для пролголимаба) и сопоставимые значения с режимом 3 мг/кг Q3W.
Результаты математического моделирования эффективности пролголимаба по показателю ЧОО в соответствии с критериями RECIST 1.1 для указанных режимов применения пролголимаба 1 мг/кг Q2W и 3 мг/кг Q3W показали, что эффективность лечения пролголимабом не является дозозависимой. Результаты, полученные в настоящем исследовании, указывают на сопоставимую эффективность при дозировании в зависимости от массы тела и при применении фиксированной дозы (см. табл. 2, рис. 2) независимо от демографических характеристик и характеристик заболевания пациента (см. табл. 1). Все пациенты, включая пациентов с высокой/низкой массой тела, достигли одинакового Ctrough независимо от режима терапии (дополнительный материал, табл. S1).
Профиль безопасности соответствовал данным, полученным в ходе других исследований пролголимаба. Частота и тяжесть НЯ были сопоставимы при обоих режимах дозирования (дополнительный материал, табл. S2, S3). Более низкая частота возникновения иоНЯ, которая наблюдалась в группе, получавшей дозу 250 мг, может быть объяснена улучшением навыков ведения пациентов, получающих иммунотерапию, с учетом времени, прошедшего между двумя исследованиями (MIRACULUM и FLAT).
Таким образом, несмотря на то что параметры ФК пролголимаба при применении дозы 250 мг Q3W были несколько выше, чем при дозе 1 мг/кг Q2W, это не оказало влияния ни на эффективность, ни на безопасность.
По сравнению с одобренными режимами лечения пролголимабом в дозе 1 мг/кг Q2W и 3 мг/кг Q3W у фиксированной дозы есть определенные преимущества: на данный момент пролголимаб доступен во флаконах 50 или 200 мг. При применении режима дозирования на основе массы тела содержимое флакона обычно вводят не полностью, а оставшийся лекарственный продукт утилизируют в соответствии с инструкциями на этикетке. В обычной клинической практике его технически возможно использовать для другого пациента, что создает проблемы с качеством и, следовательно, потенциальные проблемы в отношении безопасности, так как лекарственный препарат представляет собой источник инфекции при использовании его ненадлежащим образом вне КИ. Таким образом, при режиме дозирования пролголимаба с фиксированной дозой эффективно используется весь флакон препарата целиком и устраняется необходимость утилизировать неиспользованный препарат. Кроме того, снижается риск непреднамеренного введения неправильной дозы.
Снижение частоты введений пролголимаба удобно для врачей и пациентов, что снижает нагрузку на систему здравоохранения: сокращается частота обращений в медицинское учреждение для проведения инфузий. Например, при введении препарата один раз в две недели пациентам приходится посещать медицинское учреждение 26 раз в год, а при введении препарата один раз в три недели – всего 17 раз. Это, в свою очередь, приводит к сокращению расходов учреждений здравоохранения.
В настоящее время применение пролголимаба в дозе 250 мг Q3W одобрено для пациентов с меланомой и НМРЛ в Российской Федерации, а клинические результаты демонстрируют одинаковую эффективность и безопасность применения при этих показаниях доз 1 мг/кг Q2W, 250 мг Q3W, 3 мг/кг Q3W, проанализированных в исследованиях эффективности и безопасности пролголимаба [8, 11].
Финансирование
Клиническое исследование компании АО «Биокад».
L.V. Demidov, PhD, G.Yu. Harkevich, PhD, N.N. Petenko, PhD, V.M. Moiseenko, PhD, S.A. Protsenko, PhD, T.Yu. Semiglazova, PhD, A.V. Zimina, N.V. Kovalenko, N.V. Fadeeva, PhD, D.V. Kirtbaya, I.O. Belogortsev, D.A. Tantsyrev, S.V. Odintsova, PhD, A.I. Nesterova, PhD, K.A. Vorontsova, PhD, Yu.Yu. Makarycheva, Yu.N. Linkova, PhD, A.V. Zinkina-Orikhan, A.A. Silyutina, PhD, I.V. Sorokina, PhD, D.O. Lyaptseva, PhD, V.S. Chistyakov, PhD, A.A. Lutsky, PhD
N.N. Blokhin National Medical Research Center of Oncology
St. Petersburg Clinical Scientific and Practical Center for Specialized Types of Medical Care (Oncological)
N.N. Petrov National Medical Research Center of Oncology
Omsk Clinical Oncological Dispensary
Volgograd Regional Clinical Oncological Dispensary
Chelyabinsk Regional Clinical Center of Oncology and Nuclear Medicine
Krasnodar Oncological dispensary No. 2
Leningrad Regional Clinical Hospital
Altai Regional Oncological Dispensary
JSC Modern Medical Technologies, St. Petersburg
M.Z. Sigal Republican Clinical Oncological Dispensary
A.S. Loginov Moscow Clinical Research Center
Samara Regional Clinical Oncological Dispensary
JSC BIOCAD, St. Petersburg
Contact person: Irina V. Sorokina, sorokinaiv@biocad.ru
Prolgolimab is the first Russian PD-1 inhibitor approved for the treatment of patients with inoperable or metastatic melanoma, as well as advanced non-squamous cell lung cancer. The drug was approved in two dosage regimens calculated for body weight: 1 mg/kg once every 2 weeks and 3 mg/kg once every 3 weeks, however, the use of prolgolimab in a fixed dose seems more promising.
Methods. A multicenter, incomparable, open-label phase III study of efficacy, pharmacokinetics and safety was conducted to assess the comparability of the dosage regimen of prolgolimab 1 mg/kg once every 2 weeks and a fixed dose of 250 mg once every 3 weeks in patients who had not previously received treatment for inoperable or metastatic melanoma (BCD-100-8/FLAT, NCT05783882). The main purpose of the study: to prove the equally effective use of prolgolimab at a dose of 250 mg once every 3 weeks compared with prolgolimab at a dose of 1 mg/kg once every 2 weeks for the treatment of patients with unresectable or metastatic melanoma in terms of the frequency of objective response according to the criteria RECIST 1.1. Patients from the clinical study MIRACULUM (BCD-100-2/ MIRACULUM, NCT03269565) was a retrospective control group.
Results. 114 patients received prolgolimab at a dose of 250 mg once every 3 weeks and 61 patients received prolgolimab (Prolgo) at a dose of 1 mg/kg once every 2 weeks (retrospective control). CSR was achieved in 33.3% (95% confidence interval (CI) 24.8–42.8) of patients in the 250 mg Prolgo group compared with 32.8% (95% CI 21.3–46.0) of patients in the 1 mg/kg Prolgo group. The risk difference was 0.00, 95% CI (-0.12 – not achieved (ND)), p = 0.0082. Both therapeutic regimens had a favorable and comparable safety profile. When using a fixed dose of 250 mg once every 3 weeks, higher values of pharmacokinetic parameters were demonstrated compared to the 1 mg/kg regimen. None of the patients were found to have binding antibodies to prolgolimab.
Conclusion. The selected dosage regimen of prolgolimab with a fixed dose of 250 mg once every 3 weeks has efficacy and safety indicators comparable to the dosage regimen of 1 mg/kg once every 2 weeks.
Уважаемый посетитель uMEDp!
Уведомляем Вас о том, что здесь содержится информация, предназначенная исключительно для специалистов здравоохранения.
Если Вы не являетесь специалистом здравоохранения, администрация не несет ответственности за возможные отрицательные последствия, возникшие в результате самостоятельного использования Вами информации с портала без предварительной консультации с врачом.
Нажимая на кнопку «Войти», Вы подтверждаете, что являетесь врачом или студентом медицинского вуза.